Research Article
Comparative transcriptome to reveal the drought tolerance mechanism in hexaploid Ipomoea trifida 
2 Crop Research Institute, Sichuan Academy of Agricultural Sciences, Chengdu, 610066, Sichuan province, China
3 Ministry of Education Key Laboratory for Bio-Resource and Eco-Environment, College of Life Sciences, Sichuan University, Chengdu, 610064, Sichuan province, China
 Author
Author  Correspondence author
Correspondence author
Molecular Plant Breeding, 2017, Vol. 8, No. 11 doi: 10.5376/mpb.2017.08.0011
Received: 22 Aug., 2017 Accepted: 12 Sep., 2017 Published: 05 Dec., 2017
Peng M.F., Lin F., Feng J.Y., Zhang C., Yang S.T., He B., Pu Z.G., Tan W.F., and Li M., 2017, Comparative transcriptome to reveal the drought tolerance mechanism in Hexaploid Ipomoea trifida, Molecular Plant Breeding, 8(11): 100-112 (doi: 10.5376/mpb.2017.08.0011)
Hexaploid Ipomoea trifida (I.trifida), a wild relative of sweetpotato[Ipomoea batatas (L.) Lam.], has plentiful superior genes and a variety of anti-adversity characteristics genetically, and used as a distant hybridization materials in sweetpotato breeding. However, insufficient transcriptomic and genomic resources of hexaploid I.trifida are available for understanding the molecular mechanism underlying drought tolerance. In this study, we are the first to report a reference transcriptome using leaf, stem and root tissues under drought-stressed condition from hexaploid I.trifida, which is helpful to generate a large number of transcripts associated with drought tolerance for gene discovery. In this study, a total of 191,231, 226,262, and 171,841 contigs with a mean length of 1,100, 1,038 and 1,106 bp were obtained from the three tissues, respectively. In our assembled sequences, 62.38%, 50.08% and 47.76% of all contigs in the three tissues could be annotated to the nr database. Based on the Gene ontology (GO) analysis, 39,193, 49,997 and 34,887 contigs in leaves, roots and stems could assign with GO terms, and totaled 69 contigs up-regulated expression. In the annotated genes, we identified six transcription factors and two genes involved in drought-stress responses. Furthermore, the gene expression profiles of 15 putative genes associated with drought tolerance were analyzed to verify the reliability of the transcriptome using quantitative real-time PCR (qRT-PCR). These results provide a foundation for understanding the molecular mechanisms underlying drought stress of hexaploid I.trifida, and promoting its further utilization in sweetpotato breeding.
Background
Sweetpotato [Ipomoea batatas (L.) Lam.] is an important food crop as vegetable and food for humans and as feed for domestic animals as well as industrial materials. China is the largest producer of sweet potato in the world. Hexaploid Ipomoea trifida (I.trifida), a wild relative of sweetpotato, is considered as a potential source of resistance genes to support sweetpotato breeding (Setiawati et al., 2013). So far, hexaploid I.trifida has been confirmed as a germplasm with characteristics of improving the yields, levels of dry matter, starch, drought tolerance, resistance to certain pests and diseases (Setiawati et al., 2013). Researchers bred some novel sweetpotato cultivars with 1/8 lineage of hexaploid I.trifida (Zhang et al., 1999; Fu et al., 2000; Liu et al., 2009). However, because of its complex genome structure and size (2N=6x=90) (Hirakawa et al., 2015), the complex feature of genome has obstructed genetic studies on agronomically important characteristics such as drought tolerance and thus, the progress in genetics and genomics in this species lags far behind that in other important crop species (Aldrich and Cavender-Bares, 2011; He et al 2015a).
Drought is a major abiotic constraint to crop production especially in the subtropics and tropics regions. It is reported that drought tolerance in plants has close relationship with transcription factors and regulatory enzymes (Zhou et al., 2012). However, knowledge concerning transcription factors and regulatory enzymes related to drought-stressed hexaploid I.trifida remains deficient. There were only approximately 1,407 I.trifida expressed sequence tags (ESTs) in the National Center for Biotechnology Information EST database (http://www.ncbi.nlm.nih.gov/dbEST/dbEST_summary.html), many of which belongs to diploid I.trifida and even less to drought-stressed hexaploid I.trifida. Although some desirable genes from hexaploid I.trifida were identified and cloned, such as SGTl (a required component in some signaling pathways mediated by disease resistance (R) genes) and NBS class resistance genes (Chen et al., 2007; 2013). These limited and incomplete resources are far from enough for the genomic studies of gene discovery and analysis to stress responses. Furthermore, the transcriptome represents a comprehensive set of transcribed regions throughout the genome. Therefore, studying of transcriptome will provide important insights into the critical transcription factors and regulatory enzymes, their expression patterns, and the regulation of transcribed regions in drought stress conditions. Compared with the former methods, RNA-seq technologies are much simpler and more cost-effective, and wildly used in many plant species (Zhang et al., 2014; Robertson et al., 2010) for discovering novel transcripts and comparing gene expression, for instance, Ipomoea batatas (Tao et al., 2012), Ipomoea nil (Wei et al., 2015), Brassica napus (Trick et al., 2009), Zea mays (Li et al., 2010), and Picrorhiza kurrooa (Gahlan et al., 2012).
Drought tolerance has been observed in many plants, including wild Arachis (Brasileiro et al., 2015), Ammopiptanthus (Zhou et al., 2012), Lolium arundinaceum (Saha et al., 2015), and potato (Zhang et al., 2014). To elucidate the mechanism underlying drought stress, it is important to determine the change of gene expression level in response to this biological process. The previous researches on the response to drought stress have provided much information on stress perception and differential activation of signaling pathways. However, rare studies on gene expression profiles were reported on hexaploid I.trifida, which exhibits strong drought tolerance. Here, hexaploid I.trifida with drought-tolerance phenotypes was exposed to a prolonged period of drought stress in a greenhouse trial, and the transcriptome sequencing approach was applied based on the Illumina sequencing platform. The aims were mainly to construct databases with abundant transcripts from leaves, stems and roots of hexaploid I.trifida and mined genes with differential expression under drought treatment as well as genes involved in drought response, finally generalize the common patterns of transcriptomic responses to lasting water deficit in leaves, stems and roots. The transcriptome databases of the hexaploid I.trifida were valuable genetic resources for gapping the vacancy of genetic information and understanding the molecular mechanisms underlying drought stress. The candidate drought-responsive genes and SSRs identified in this study can be applied to molecular breeding and selecting of its filial generation for cultivated sweetpotato with enhanced drought tolerance.
1 Results
1.1 Sequencing and de novo assembly of hexaploid I.trifida transcriptome
The two libraries (drought-treated and control) with three tissues were sequenced respectively using Illumina HiSeq2000. To investigate genes involved in drought tolerance that might be expressed in different vegetative organs, samples collected from roots, stems and leaves were used for RNA-Seq analysis. After filtering, 101.1M, 105.85M and 95.37M pair-end (PE) of 90 bp clean reads were obtained from leaves, roots and stems samples respectively (Table 1). The filtered reads were de novo assembled by Trinity (kmer length = 25). The assembly results were consisted of 191,231, 226,262, and 171,841 contigs. The mean length of these contigs were 1,100, 1,038 and 1,106 bp and the N50 value were 1,850, 1,802, 1,833 bp. The size distribution of contigs lengths is shown in Figure 1.
.jpg) Table 1 Throughput and quality of RNA-Seq of hexaploid I. trifida transcriptome |
.jpg) Figure 1 Length distributions of hexaploid I. trifida transcriptome |
To evaluate the quality of the assembly, the Trinity utility were used to predict potential ORFs from the reconstructed contigs, resulting in the prediction of 100,205 (52.39%), 115,570 (51.08%), and 90,397 (52.61%) best candidate ORFs from leaves, roots and stems respectively. Of these contigs with CDS, there were 45.12%, 41.19% and 45.86% of them containing ORFs ≥ 900 bp. These results suggested that most of these contigs were protein-encoding transcripts and indicated the satisfactory of de novo assembly.
1.2 Functional annotation and classification
The assembly contigs were annotated through searching specific databases based on sequence similarity. In this study, all of the contigs were matched to the sequences in the non-redundant (nr) database using BLASTX with a cut off e-value of 10-5. In leaves, a total of 105,315 contigs (55.07% of all contigs) returned a significant BLAST result and 113,302 contigs (50.08% of all contigs) in roots, 82,065 contigs (47.76% of all contigs) in stems could be annotated to the nr database.
To further annotation, the contigs homologous to known sequences in nr were compared with GO database using Blast2GO. In total, 39,193, 49,997 and 34,887 contigs in leaves, roots and stems were assigned with GO terms. The gene functional classification by GO analysis showed that the largest GO terms in leaves were “cell”, “organelle”, “metabolic process”, “cellular process”, “catalytic activity” and “binding”. The largest GO terms and majorities of GO terms in other two tissue samples were similar, while there were some differences in “synapse”, “chemorepellent” and “locomotion” among the three tissues (Figure 2).
.jpg) Figure 2 The gene ontology (GO) classification of hexaploid I. trifida transcriptome Note: 1~14: Cellilar component; 1: cell; 2: cell part; 3: envelope; 4: extracellular region; 5: extracellular region part; 6: macromolecular complex; 7: membrane-enclosed lumen; 8: organelle; 9: organelle part; 10: symplast; 11: synapse; 12: synapse part; 13: virion; 14: virion part; 15~29: Molecular function; 15: antioxidant; 16: auxiliary transport protein; 17: binding; 18: catalytic; 19: chemorepellent; 20: electron carrier; 21: enzyme regulator; 22: metallochaperone; 23: molecular transducer; 24: nutrient reservoir; 25: protein tag; 26: structural molecule; 27: transcription regulator; 28: translation regulator; 29: transporter; 30~52: Biological process; 30: anatomical structure formation; 31: biological adhesion; 32: biological regulation; 33: cell killing; 34: cellular component biogenesis; 35: cellular component organization; 36: cellular process; 37: death; 38: developmental process; 39: establishment of localization; 40: growth; 41: immune system process; 42: localization; 43: locomotion; 44: metabolic process; 45: multi-organism process; 46: multicellular organismal process; 47: pigmentation; 48: reproduction; 49: reproductive process; 50: response to stimulus; 51: rhythmic process; 52: viral reproduction |
To make further understanding of the transcriptome data, pathway analysis with KEGG mapping of the hexaploid I.trifida transcriptome were carried out. Totally 17,590, 18,529 and 16,517 contigs in leaves, roots and stems were identified with pathway annotation, and functionally assigned to 338 KEGG pathways. Figure 3 summarizes the top 10 represented biological pathways. The most highly represented pathways were ‘Biosynthesis of secondary metabolites’ and ‘Microbial metabolism in diverse environments’. Pathways associated with ‘Ribosome’, ‘Spliceosome’ and ‘Biosynthesis of amino acids’ were also higly represented (Figure 3). These results suggested that there were not obvious difference presented on the function of genes in the three tissues.
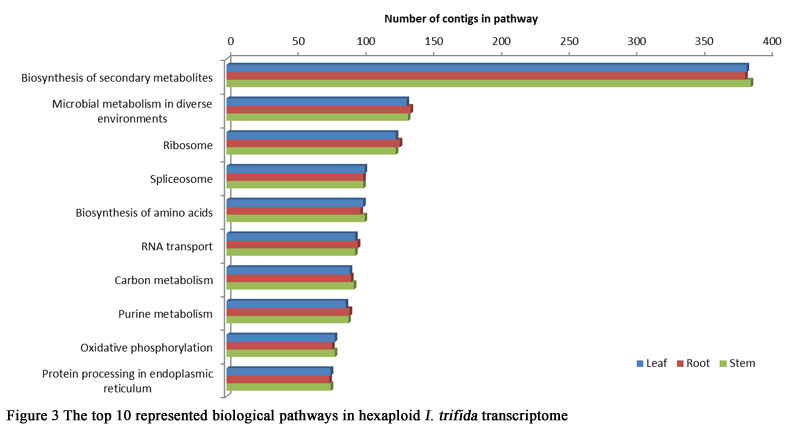 Figure 3 The top 10 represented biological pathways in hexaploid I. trifida transcriptome |
1.3 Characterization of SSRs
The transcript/EST-based markers are important resource for determining functional genetic variation. In order to evaluate the usefulness of this database for marker development, all of the assembled sequences from three tissues were mined for SSRs. Using the MISA Perl script, a total of 26,283, 29,926 and 23,320 SSRs were identified in 22,743, 25,788 and 20,016 sequences, respectively from leaves, roots and stems (Table 2). The di-nucleotide SSRs represented the largest fraction of SSRs followed by tri-nucleotide in the three tissues. In addition, there was no hexa-nucleotide SSRs identified in any of the three tissues. Furthermore, there were 1,066, 1,318 and 1,040 SSRs leaves, roots and stems presented in compound formation. The SSRs mining from this database will be valuable resource for genetic manipulations in hexaploid I.trifida.
.jpg) Table 2 Summary of SSRs identification from hexaploid I. trifida transcriptome |
1.4 Comparison of transcriptome patterns between control and drought-treated hexaploid I.trifida
To determine differentially expressed genes between drought-treated and control, the log2 (fold change) of drought treatment versus control FPKM values for each gene were counted. On the basis of the applied criteria, a total of 5,389 genes were differentially expressed in leaves of hexaploid I.trifida, including 18 up- and 5,371 down-regulated, by drought-treated. This is similar with the number of genes affected by drought in roots and stems, which accounted for 6,537 and 5,644 differentially expressed genes (DEGs). Of the DEGs in roots, 28 were up-regulated and 6,509 were down-regulated, while there were 20 DEGs up-regulated and 5,624 DEGs down-regulated. The GO classification of the differentially expressed contigs in leaves indicated that the up-regulated DEGs were annotated to few GO terms: “cell”, “organelle”, “response to stimulus”, “cellular process”, “binding” and “catalytic activity”, while the down-regulated DEGs were annotated to 51 GO terms (Figure 4). The up-regulated DEGs in roots could match more GO terms than those in leaves, but less numbers in each matched GO terms. The most abundant GO terms of up-regulated DEGs in stems was “metabolic process”, followed by “cell”, “organelle” and “binding” (Figure 5). Of the down-regulated DEGs in stems, except the GO terms same with up-regulated DEGs, the largest GO terms were “macromolecular complex”, “localization”, “cellular component organization” and “multicellular organismal process” (Figure 6).
.jpg) Figure 4 The gene ontology (GO) classification of differentially expressed genes in leaves Note: 1~12: Cellilar component; 1: cell; 2: cell part; 3: envelope; 4: extracellular region; 5: extracellular region part; 6: macromolecular complex; 7: membrane-enclosed lumen; 8: organelle; 9: organelle part; 10: symplast; 11: virion; 12: virion part; 13~25: Molecular function; 13: antioxidant; 14: binding; 15: catalytic; 16: electron carrier; 17: enzyme regulator; 18: metallochaperone; 19: molecular transducer; 20: nutrient reservoir; 21: protein tag; 22: structural molecule; 23: transcription regulator; 24: translation regulator; 25: transporter; 26~47: Biological process; 26: anatomical structure formation; 27: biological adhesion; 28: biological regulation; 29: cellular component biogenesis; 30: cellular component organization; 31: cellular process; 32: death; 33: developmental process; 34: establishment of localization; 35: growth; 36: immune system process; 37: localization; 38: locomotion; 39: metabolic process; 40: multi-organism process; 41: multicellular organismal process; 42: pigmentation; 43: reproduction; 44: reproductive process; 45: response to stimulus; 46: rhythmic process; 47: viral reproduction |
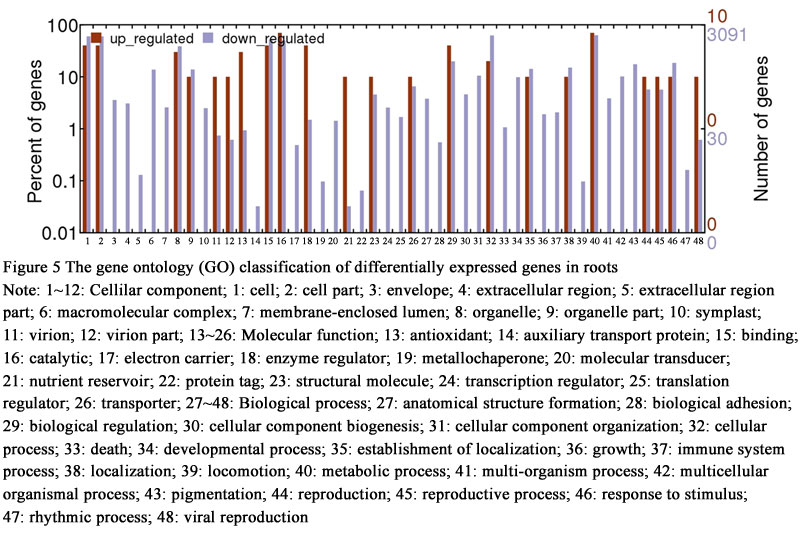 Figure 5 The gene ontology (GO) classification of differentially expressed genes in roots Note: 1~12: Cellilar component; 1: cell; 2: cell part; 3: envelope; 4: extracellular region; 5: extracellular region part; 6: macromolecular complex; 7: membrane-enclosed lumen; 8: organelle; 9: organelle part; 10: symplast; 11: virion; 12: virion part; 13~26: Molecular function; 13: antioxidant; 14: auxiliary transport protein; 15: binding; 16: catalytic; 17: electron carrier; 18: enzyme regulator; 19: metallochaperone; 20: molecular transducer; 21: nutrient reservoir; 22: protein tag; 23: structural molecule; 24: transcription regulator; 25: translation regulator; 26: transporter; 27~48: Biological process; 27: anatomical structure formation; 28: biological adhesion; 29: biological regulation; 30: cellular component biogenesis; 31: cellular component organization; 32: cellular process; 33: death; 34: developmental process; 35: establishment of localization; 36: growth; 37: immune system process; 38: localization; 39: locomotion; 40: metabolic process; 41: multi-organism process; 42: multicellular organismal process; 43: pigmentation; 44: reproduction; 45: reproductive process; 46: response to stimulus; 47: rhythmic process; 48: viral reproduction |
.jpg) Figure 6 The gene ontology (GO) classification of differentially expressed genes in stems Note: 1~12: Cellilar component; 1: cell; 2: cell part; 3: envelope; 4: extracellular region; 5: extracellular region part; 6: macromolecular complex; 7: membrane-enclosed lumen; 8: organelle; 9: organelle part; 10: symplast; 11: virion; 12: virion part; 13~26: Molecular function; 13: antioxidant; 14: auxiliary transport protein; 15: binding; 16: catalytic; 17: electron carrier; 18: enzyme regulator; 19: metallochaperone; 20: molecular transducer; 21: nutrient reservoir; 22: protein tag; 23: structural molecule; 24: transcription regulator; 25: translation regulator; 26: transporter; 27~48: Biological process; 27: anatomical structure formation; 28: biological adhesion; 29: biological regulation; 30: cellular component biogenesis; 31: cellular component organization; 32: cellular process; 33: death; 34: developmental process; 35: establishment of localization; 36: growth; 37: immune system process; 38: localization; 39: locomotion; 40: metabolic process; 41: multi-organism process; 42: multicellular organismal process; 43: pigmentation; 44: reproduction; 45: reproductive process; 46: response to stimulus; 47: rhythmic process; 48: viral reproduction |
1.5 Identification and expression profiling of genes involved in drought response
Transcription factors and regulatory enzymes were critical elements of drought stress tolerance. In some plants, many of these genes previously have been confirmed and identified as key regulators of drought responses. Plants could response to drought through two pathways: abscisic acid (ABA)-dependent pathway and ABA-independent pathway. In the current study, two genes and six transcription factors involved in drought response were identified in hexaploid I.trifida transcriptome, including DREB (dehydration-responsive element-binding protein), MYB (myeloblastosis oncogenes), MYC, NAC (nascent polypeptide-associated complex), HD-ZIP (Homeodomain-Zipper), RD22 (Dehydration responsive protein 22), ERD1 (Early Responsive to Dehydration 1) and bZIP (basic leucine zipper) protein. Transcription factors MYB, MYC and bZIP acted in the ABA-dependent pathway but only RD22 was expressed in the downstream, while NAC, HD-ZIP and DREB worked in the ABA-independent pathway and ERD1 was expressed in the downstream (Figure 7).
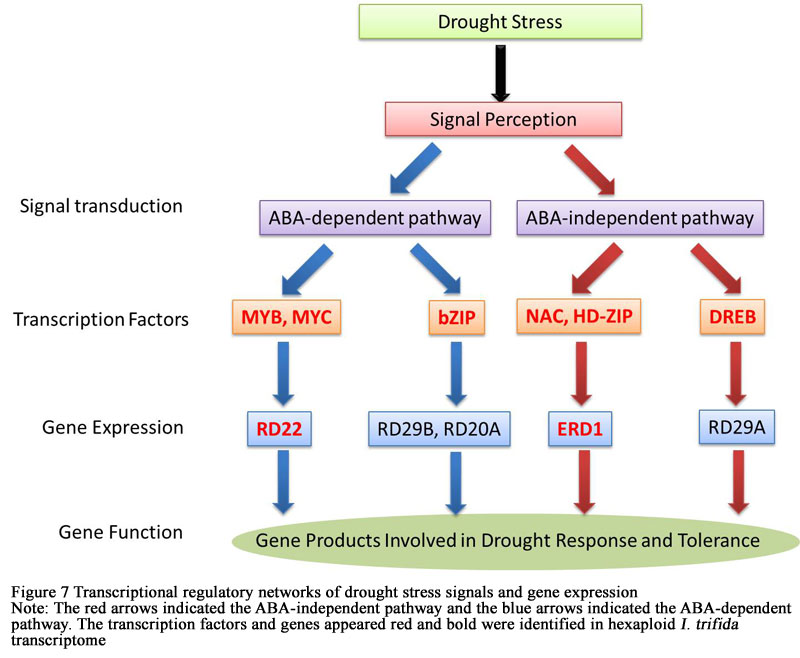 Figure 7 Transcriptional regulatory networks of drought stress signals and gene expression Note: The red arrows indicated the ABA-independent pathway and the blue arrows indicated the ABA-dependent pathway. The transcription factors and genes appeared red and bold were identified in hexaploid I. trifida transcriptome |
To verify the reliability and reproducibility of the transcriptome analysis results, we selected randomly 10 up-regulated genes for qRT-PCR validation. Templates were from the control and drought-treated leaves, roots and stems RNA samples. The expression profiles of the candidate contigs revealed by qRT-PCR analysis were consistent with the results of the transcriptome, confirming our transcriptome analysis (Figure 8; Table 3).
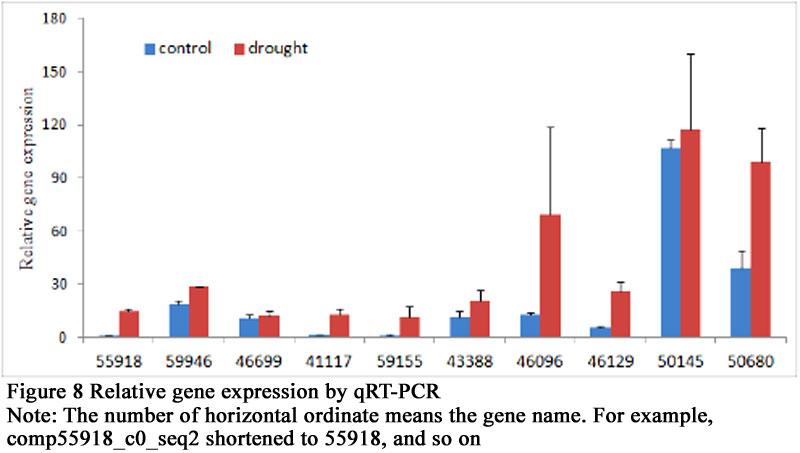 Figure 8 Relative gene expression by qRT-PCR Note: The number of horizontal ordinate means the gene name. For example, comp55918_c0_seq2 shortened to 55918, and so on |
.jpg) Table 3 The primer of 10 selected transcripts |
2 Discussion
In 600-700 kinds of Ipomoea genus plants, I.trifida is considered most likely the ancestors of the sweetpotato, and hexaploid I.trifida has closest wild relative with sweetpotato (Hirakawa et al., 2015; Roullier et al., 2013). So, hexaploid I.trifida is considered as resistance germplasm in sweetpotato distant hybridization. Investigation of the gene expression regulation network under drought stress will be helpful to understand the physiological and biochemical adaptation process in hexaploid I.trifida. This kind of mechanism and SSRs for drought stress could be also used in the selecting of its filial generation. Drought is a worldwide problem, constraining crop production seriously and recent global climate change has made this situation more serious. Investigating the drought-tolerance mechanisms in hexaploid I.trifida could facilitate a better understanding of the genetic bases of drought tolerance, and be beneficial to the effective use of genetic and genomic approaches for improvement of sweetpotato breeding and production. The adaptive ability of sweetpotato which contained wild blood, such as for drought stress, is much more than ordinary sweetpotato cultivars. This is the reason why sweetpotato breeders selected the wild relatives as parent plant, such as hexaploid I.trifida. And more important is that some sweetpotato cultivars were bred through hexaploid I.trifida (Zhang et al 1999; Fu et al., 2000; Liu et al., 2009). Recently, the diploid I.trifida used in breeding and made important progress (Cao et al., 2014). But the hexaploid I.trifida cross with sweetpotato more directly.
In the present study, RNA-seq technology were used for transcriptome profiling to sequence mRNA extracted from leaves, stems and roots under control and drought condition. Through this procedure, we obtained three transcriptome database including leaves, roots and stems as the first step of our endeavor to provide a clear insight into the molecular mechanism of drought tolerance in hexaploid I.trifida. This method could be avoid the false negatives and difficult to interpret patterns of differential expression by composite extractions from different tissues or assembly transcriptomic data using sequencing reads from different tissues (He et al., 2015b; Hou et al., 2011; Fu and He, 2012; Wang et al., 2013). Here, we did not use the flower, that’s because the hexaploid I.trifida not formed the flower bud under a sustained severe drought stress.
From the clean reads, 191,231, 226,262, and 171,841 contigs were assembled from leaves, roots and stems of hexaploid I.trifida. When annotated to nr database, many contigs could not be matched the known non-redundant protein, especially in the stems (52.24%). Compared with previous transcriptome study on Ipomoea batatas (Schafleitner et al., 2010; Wang et al., 2010; Tao et al., 2012, Xie et al., 2012; Ma et al., 2016), more transcripts in hexaploid I.trifida had significant BLASTX hits to the nr database than these in cultivar ‘Xushu18’ (40.42%) (Tao et al., 2012) and less than variety ‘Georgia Jet’ (72.48%) (Firon et al., 2013), and it also less than diploid I.trifida (76.68%) (Cao et al., 2016). This may be because of the characters with hexaploid genome and wild. But our results may provide more genetic information for Ipomoea. In addition, the results of functional annotation and classification in the three tissues revealed that there was little discrepancy on the function of genes most annotated among the three tissues. For example, when annotated to the GO database, the largest GO terms in the three tissues were “cell”, “organelle”, “metabolic process”, while mapped to the KEGG database, the most highly represented pathways in the three tissues were ‘Biosynthesis of secondary metabolites’ and ‘Microbial metabolism in diverse environments’. The number and function of differentially expressed genes among the three tissues were even no significant difference. There results may suggest that the expression of genes in hexaploid I.trifida regardless of drought treated would not change much among the three tissues.
Drought is a worldwide problem, constraining global crop production seriously. Investigating the drought-tolerance mechanisms in I.trifida could facilitate a better understanding of the genetic bases of drought tolerance, and be beneficial to facilitate the effective use of genetic and genomic approaches for improvement of crop production improvement (Ahmed et al., 2015). It is well-established that, under drought stress, drought triggers the production of the phytohormone abscisic acid (ABA), which in turn causes the closure of leaf stomata and induces expression of stress-related genes (Waseem et al., 2011). Several drought-inducible genes are induced by exogenous ABA treatment, whereas others are not affected. Indeed, evidences demonstrated that the presence of both ABA-independent and ABA-dependent regulatory systems govern drought-inducible gene expression. At least two ABA dependent and two ABA independent signal transduction pathways exist in drought-stress responses (Shinozaki and Yamaguchi-Shinozaki, 2007). bZIP families function as transcription factors involved in ABA-dependent pathway while MYB2 and MYC2 function as transcription factors in ABA-inducible gene expression of the RD22 gene. In one of the ABA-independent pathways, DREBs are mainly involved in the regulation of genes by drought stress which resulted in the expression of RD29A. In the other ABA-independent pathways, the NAC and HD-ZIP transcription factors are involved in ERD1 gene expression. In the present study, we identified transcription factors involved in four signal transduction pathways in drought-stress responses. However, there were only two genes expressed with the existence of these transcription factors, one in ABA-dependent pathway (RD22) and one in ABA-independent pathway (ERD1) (Figure 4). In addition, our results showed that there were no significant differences on the expression of these transcription factors and genes between the control and drought-stress groups. The reason may be that these transcription factors and genes in hexaploid I.trifida involved in drought-stress responses were constitutive expression. Therefore, we could get the conclusion that only two pathways existed in hexaploid I.trifida related to drought-stress responses, although there were transcription factors existed in four signal transduction pathways.
3 Materials and Methods
3.1 Sample preparation and RNA Isolation
Plantlets of hexaploid I.trifida were transferred to pots (one per pot) with six replicates each in greenhouse under natural light conditions. Plants were irrigated one times each day in the first 20 days. When plants were survived completely, drought stresses were applied by stopping irrigation in treatment plot, meanwhile, the control plot were irrigated continuously. Leaves, stems and roots were collected from treatment and control on 60 days after drought onset, on each time-point. These materials were sampled from six plants and pooled together, immediately frozen in liquid nitrogen and stored at −80 °C prior to RNA extraction.
Total RNA was isolated directly from frozen materials using Trizol Reagent (Invitrogen) according to the manufacturer’s instructions. A DNase (TaKaRa) treatment was also applied to remove any residual genomic DNA. RNA purity were checked using the NanoPgotometer spectrophotometer (IMPLEN, CA, USA) and RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
3.2 Library construction and Illumina sequencing
The cDNA library was constructed using an cDNA Sample Preparation Kit (Illumina) following the manufacturer’s instructions. The total RNA was collected from each control and treatment tissues, and mRNA was enriched and purified by the ploy-T oligo-attached magnetic beads, respectively. The mRNA was fragmented using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X) subsequently. The cleaved RNA fragments were then primed with random hexamers and submitted to the synthesis of the first-strand and second-strand cDNAs. The cDNAs fragments processed for end repair and finally ligated to paired end adaptors. Then, the cDNA fragments with 150-200 bp size were selected by agarose gel electrophoresis and enriched by PCR amplification. PCR products were purified (AMPure XP system) and library quality was accessed on the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Finally, the library preparations were sequenced on an Illumina HiSeq2000 sequencing platform in Beijing Novogene Bioinformatics Technology Co., Ltd (China). The transcriptome datasets are available in the Illumina instrument software.
3.3 Data filtering and de novo assembly
After sequencing was completed, raw reads (produced from the sequencing machines) that contained adapters, reads containing unknown sequences ‘N’ with a rate more than 5%, and low-quality bases which were identified based on CycleQ30 (corresponding to a 0.1 % sequencing error rate) were removed before assembly. The remaining Illumina sequences were first assessed by FastQC (v0.10.0) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and then de novo assembled using Trinity_release_20131110 under default parameters (Haas et al., 2013). In order to evaluate the quality of assembly, the initial assembly quality was evaluated by custom perl scripts using the following criterion: total number of contigs, maximum contig length, mean contig length and N50 (He et al., 2015b). Moreover, to further investigate the assembly quality, potential coding regions within contigs were analyzed with the Trinity utility (transcripts_to_best_scoring_ORFs.pl) (Pang et al., 2013).
3.4 Functional annotation
For annotation, all contigs were first searched against NCBI non-redundant protein (NR) database using the BLASTX algorithm with an E value cut-off of 1e-5. Then gene ontology (GO) analysis was conducted on the annotated sequences through localized Blast2GO (Conesa et al., 2005). Gene ontology classification was achieved using WEGO [http://wego.genomics.org.cn/cgi-bin/wego/index.pl]. To further enrich the pathway annotation and to identify the BRITE functional hierarchies, sequences were also submitted to the KEGG Automatic Annotation Server (KAAS), and the single-directional best hit information method was selected (Moriya et al., 2007).
3.5 Differential expression analysis
To investigate the expression profiles of each contig in different samples, RSEM was used for abundance estimation for transcriptome assemblies. After executing RSEM accordingly, FPKM (fragments per feature kilobase per million reads mapped) for each sample was calculated as the expression levels of each contig, which could eliminate the influence of gene length on the calculation of gene expression (Mortazavi et al., 2008).
Based on the abundance estimation described above, we then determined the statistically significant differences in gene expression for individual transcripts between samples (Audic and Claverie, 1997). The identification of differentially expressed transcripts relies on using the EdgeR Bioconductor package and FDR (False Discovery Rate) control method was used to correct the results for p value. To extract those transcripts that are most differentially expressed, the ratio of FPKMs was used as well to calculate the fold-change according to their patterns of differential expression across the samples. In the current study, the differentially expressed genes (DEGs) were screened with the value of FDR≤0.001 and the threshold of log2fold-change (log2FC)≥1 (Pang et al., 2013).
3.6 Identification of SSRs
All assembled sequences were searched for the presence of simple sequence repeats (SSRs) using the MISA tool (MIcroSAtellite; http://pgrc.ipk-gatersleben.de/misa/), which identifies both perfect and compound repeats. SSRs with motifs were searched ranging from mono- to hexa-nuclotides in size and with a minimum length of 18bp. The parameters were adjusted for identification of perfect mono-, di-, tri-, tetra-, penta-, and hexanuclotide motifs with a minimum of 15, 9, 6, 5, 4, and 3 repeats, respectively. Adjacent microsatellites≤10 nt apart were considered compound repeats (Pang et al., 2013).
3.7 Quantitative real-time PCR analysis
The total RNA isolated by RNAprep pure Plant Kit (TIANGEN BIOTECH, BEIJING) were used to synthesize the first-strand cDNA by oligo (dT)18 and 6 random primers. Quantitative real-time reverse transcription - PCR (QRT-PCR) was carried out using diluted cDNA and SsoFastTM EvaGreen Superemix (Bio-Rad, USA) in the Bio-Rad iCycler MyiQ Real-Time PCR Systems. The qRT-PCR cycle profile included one cycle of 95℃ for 30 s, followed by 40 cycles of 95℃ for 5 s, 55℃ for 30 s, and a final Melt curve profile (65-95℃). Quantification was determined with comparative CT method. Each data represented the average of three repeats.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31471448, 31101119), Sichuan Financial Innovation Project, China (2015LWJJ-001, 2016QNJJ-001, 2015KXJJ-002, 2015JSCX-007, 2016ZYPZ-005), Sichuan Breeding Cooperation Project, China (2016NYZ0032), and the earmarked fund for China Agriculture Research System (CARS-11-B-04).
Authors' contributions
M.P. and F.L. carried out the qRT-PCR analysis, participated in assembled the transcripts and drafted the manuscript. J.F. carried out the SSRs analysis. C.Z., S.Y. and B.H participated in the transcripts analysis. Z.P. and W.T. prepared the plant material and RNA. M.L. conceived and design of the study. All authors read and approved the final manuscript.
Data availability
The full data sets of hexaploid Ipomoea trifida have been submitted to NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra/) under Biosample Accession SAMN06833858, Bioproject: PRJNA384881.
References
Ahmed I.M., Nadira U.A., Bibi N., Zhang G., and Wu F., 2015, Tolerance to combined stress of drought and salinity in Barley, In: Combined Stresses in Plants, Springer International Publishing Switzerland, pp.93-121
https://doi.org/10.1007/978-3-319-07899-1_5
Audic S., and Claverie J.M., 1997, The significance of digital gene expression profiles, Genome Research, 7(10): 986-995
https://doi.org/10.1101/gr.7.10.986
Brasileiro A.C., Morgante C.V., Araujo A.C., Leal-Bertioli S.C.M., Sliva A.K., Martins A.C.Q., Vinson C.C., Santos C.M.R., Bonfim O., Togawa R.C., Saraiva M.A.P., Bertioli D.J., and Guimaraes P.M., 2015, Transcriptome profiling of wild Arachis from water-limited environments uncovers drought tolerance candidate genes, Plant Molecular Biology Reporter, 33: 1876-1892
https://doi.org/10.1007/s11105-015-0882-x
Cao Q.H., Li A., Chen J.Y., Sun Y., Tang J., Zhang A., Zhou Z.L., Zhao D.L., Ma D.F., and Gao S., 2016, Transcriptome sequencing of the sweet potato progenitor (Ipomoea Trifida (H.B.K.) G. Don.) and discovery of drought tolerance genes, Tropical Plant Biology, 9(2): 63-72
https://doi.org/10.1007/s12042-016-9162-7
Cao Q.H., Tang J., Li A., Gruneberg W., Huamani K., and Ma D., 2014, Ploidy level and molecular phylogenic relationship among novel Ipomoea interspecific hybrids, Czech Journal of Genetics and Plant Breeding, 50: 32-38
Chen G.S., Bai J., Xia N., Zhou Y.F., and Pan D.R., 2013, Cloning and characterization of ItSGT1 gene from Ipomoea trifida, Acta Botanica Boreali-Occidentalia Sinica, 33(2): 223-228 (in English, chinese abstract)
Chen G.S., Pan D.R., Zhou Y.F., and Gao L.H., 2007, Cloning and analysis of NBS class resistance gene analogues in Ipomoea trifida, 27(9): 1728-1734 (in English, chinese abstract)
Conesa A., Götz S., García-Gómez J.M., Terol J., Talón M., and Robles M., 2005, Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research, Bioinformatics, 21(18): 3674-3676
https://doi.org/10.1093/bioinformatics/bti610
Firon N., LaBonte D., Villordon A., Kfir Y., Solis J., Lapis E., Perlman T.S., Doron-Faigenboim A., Hetzroni A., Althan L., and Nadir L.A., 2013, Transcriptional profiling of sweetpotato (Ipomoea batatas) roots indicates down-regulation of lignin biosynthesis and up-regulation of starch biosynthesis at an early stage of storage root formation, BMC Genomics, 14: 460
https://doi.org/10.1186/1471-2164-14-460
Fu B., and He S., 2012, Transcriptome analysis of silver carp (Hypophthalmichthys molitrix) by paired-end RNA sequencing, DNA Research, 19(2): 131-142
https://doi.org/10.1093/dnares/dsr046
Fu Y.F., Zhang Q.T., Yang C.X., Zhang L.Y., and Qiu R.L., 2000, Studies on the breeding of “Yusu 297”, a newly-evolved variety of sweet potato, Journal of Southwest China Normal University (Natural Science), 25(6): 694-699 (in Chinese, english abstract)
Gahlan P., Singh H.R., Shankar R., Sharma N., Kumari A., Chawla V., Ahuja P.S., Kumar S., 2012, De novo sequencing and characterization of Picrorhiza kurrooa transcriptome at two temperatures showed major transcriptome adjustments, BMC Genomics, 13: 126
https://doi.org/10.1186/1471-2164-13-126
Haas B.J., Papanicolaou A., Yassour M., et al., 2013, De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis, Nature Protocols, 8(8): 1494-1512
https://doi.org/10.1038/nprot.2013.084
He B., Tao X., Gu Y., Wei C., Cheng X., Xiao S., Cheng Z., and Zhang Y., 2015a, Transcriptomic analysis and the expression of disease-resistant genes in Oryza meyeriana under native condition, PloS One, 10(12): e0144518
https://doi.org/10.1371/journal.pone.0144518
He B., Zhao S., Chen Y., Cao Q., Wei C., Cheng X., and Zhang Y., 2015b, Optimal assembly strategies of transcriptome related to ploidies of eukaryotic organisms, BMC Genomics, 16(1): 65
https://doi.org/10.1186/s12864-014-1192-7
Hirakawa H., Okada Y., Tabuchi H., et al., 2015, Survey of genome sequences in a wild sweet potato, Ipomoea trifida (HBK) G. Don, DNA Research, 22(2): 171-179
https://doi.org/10.1093/dnares/dsv002
Hou R., Bao Z., Wang S, Su H., Li Y., Du H., Hu J., Wang S., and Hu X., 2011, Transcriptome sequencing and de novo analysis for Yesso scallop (Patinopecten yessoensis) using 454 GS FLX, PLoS One, 6(6): e21560
https://doi.org/10.1371/journal.pone.0021560
Kole C., 2011, Wild crop relatives: genomic and breeding resources, Springer-Verlag Berlin Heidelberg
https://doi.org/10.1007/978-3-642-21102-7
Li P., Ponnala L., Gandotra N., et al., 2010, The developmental dynamics of the maize leaf transcriptome, Nature Genetics, 42(12): 1060-1067
https://doi.org/10.1038/ng.703
Liu L.F., Ma Z.M., and Zhang S.S., 2009, Breeding of new sweet potato variety Jishu99 and its high yield physiological characters, Journal of Hebei Agricultural Sciences, 13 (7):6 -8 (in Chinese, english abstract)
Ma P, Bian X, Jia Z, Guo X, and Xie Y., 2016, De novo sequencing and comprehensive analysis of the mutant transcriptome from purple sweet potato (Ipomoea batatas L.), Gene, 575(2 Pt 3): 641-649
https://doi.org/10.1016/j.gene.2015.09.056
Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M., 2007, KAAS: an automatic genome annotation and pathway reconstruction server, Nucleic Acids Research, 35 (Web Server issue): W182-W185
Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B., 2008, Mapping and quantifying mammalian transcriptomes by RNA-Seq, Nature Methods, 5(7): 621-628
https://doi.org/10.1038/nmeth.1226
Pang T., Ye C.Y., Xia X., Yin W., 2013, De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using Illumina/Solexa, BMC Genomics, 14(1): 488
https://doi.org/10.1186/1471-2164-14-488
Robertson G., Schein J., Chiu R., et al., 2010, De novo assembly and analysis of RNA-seq data, Nature methods 7(11): 909-912
https://doi.org/10.1038/nmeth.1517
Roullier C., Duputié A., Wennekes P., et al., 2013, Disentangling the origins of cultivated sweet potato (Ipomoea batatas (L.) Lam.), PLoS One, 8(5): e62707
https://doi.org/10.1371/journal.pone.0062707
Saha M.C., Talukder S., Azhaguvel P., Mukhergee S., and Chekhovskiy K., 2015, Deciphering drought tolerance in tall fescue [Lolium arundinaceum (Schreb.) Darbysh.], Molecular Breeding of Forage and Turf: Springer pp.1-7
Schafleitner R., Tincopa L.R., Palomino O., et al., 2010, A sweetpotato gene index established by de novo assembly of pyrosequencing and Sanger sequences and mining for gene-based microsatellite markers, BMC Genomics, 11: 604
https://doi.org/10.1186/1471-2164-11-604
Setiawati T., and Karuniawan A., 2013, Diversity analysis of tubered-bearing Ipomoea trifida (H.B.K.) G. Don. originated from Citatah West Java Indonesia based onchromosome traits, Scientific Bulletin (Series F: Biotechnologies), 17: 35-38
Shinozaki K, and Yamaguchi-Shinozaki K., 2007, Gene networks involved in drought stress response and tolerance, Journal of Experimental Botany, 58(2): 221-227
https://doi.org/10.1093/jxb/erl164
Tao X., Gu Y.H., Wang H.Y., Zheng W., Li X., and Zhao C.W., 2012, Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam.], PLoS One, 7(4): e36234
https://doi.org/10.1371/journal.pone.0036234
Trick M., Long Y., Meng J., and Bancroft I., 2009, Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing, Plant Biotechnology Journal., 7(4): 334-346
https://doi.org/10.1111/j.1467-7652.2008.00396.x
Wang Y., Hua W., Wang J., Hannoufa A., Xu Z., and Wang Z., 2013, Deep sequencing of Lotus corniculatus L. reveals key enzymes and potential transcription factors related to the flavonoid biosynthesis pathway, Molecular Genetics and Genomics, 288(3-4): 131-139
https://doi.org/10.1007/s00438-013-0736-x
Wang Z., Fang B., Chen J., Zhang X., Luo Z., Huang L., Chen X., and Li Y., 2010, De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas), BMC Genomics, 11: 726
https://doi.org/10.1186/1471-2164-11-726
Waseem M., Ali A., Tahir M., et al., 2011, Mechanism of drought tolerance in plant and its management through different methods, Continental Journal of Agricultural Science, 5(1): 10-25
Wei C., Tao X., Li M., He B., Yan L., Tan X., and Zhang Y., 2015, De novo transcriptome assembly of Ipomoea nil using Illumina sequencing for gene discovery and SSR marker identification, Molecular Genetics and Genomics, 290(5): 1873-1884
https://doi.org/10.1007/s00438-015-1034-6
Xie F., Burklew C.E., Yang Y., Liu M., Xiao P., Zhang B., and Qiu D., 2012, De novo sequencing and a comprehensive analysis of purple sweet potato (Ipomoea batatas L.) transcriptome, Planta, 236: 101-113
https://doi.org/10.1007/s00425-012-1591-4
Zhang N., Liu B., Ma C., Zhang G., Chang J., Si H., Wang D., 2014, Transcriptome characterization and sequencing-based identification of drought-responsive genes in potato, Molecular Biology Reports, 41(1): 505-517
https://doi.org/10.1007/s11033-013-2886-7
Zhang Q.T., Fu Y.F., Yang C.X., Zhang L.Y., Qiu R.L., 1999, Studies on the Breeding of “Yusu 303”, a newly-evolved variety of sweet potato, Journal of Southwest China Normal University (Natural Science), 24(6): 678-684 (in Chinese, english abstract)
Zhou Y., Gao F., Liu R., Feng J., and Li H., 2012, De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus, BMC Genomics, 13(1): 266



. PDF(347KB)
. FPDF(win)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Meifang Peng
. Feng Lin
. Junyan Feng
. Cong Zhang
. Songtao Yang
. Bin He
. Zhigang Pu
. Wenfang Tan
. Ming Li
Related articles
. Ipomoea trifida
. Hexaploid
. Drought stress
. Transcriptome
. qRT-PCR, Sweetpotato
Tools
. Email to a friend
. Post a comment