Research Article
Principle and Strategy of DNA Fingerprint Identification of Plant Variety 
2 Institute of Biotechnology, Jiangsu Academy of Agricultural Sciences, Nanjing, 210014, China
3 Beijing TodaySoft Inc., Beijing, 100083, China
4 Institute of Cotton Research of CAAS, Anyang, 455000, China
5 National Agricultural Technical Extension and Service Center, Beijing, 100125, China
* These authors contributed equally to this work
 Author
Author  Correspondence author
Correspondence author
Molecular Plant Breeding, 2019, Vol. 10, No. 11 doi: 10.5376/mpb.2019.10.0011
Received: 08 Jun., 2019 Accepted: 28 Jul., 2019 Published: 11 Aug., 2019
Wang F.G., Tian H.L, Yi H.M., Zhao H., Huo Y.X., Kuang M., Zhang L.K., Lv Y.D., Ding M.Q., and Zhao J.R., 2019, Principle and strategy of dna fingerprint identification of plant variety, Molecular Plant Breeding, 10(11): 81-92 (doi: 10.5376/mpb.2019.10.0011)
Plant variety identification has profound meanings to ensure seed quality and food safety. DNA fingerprinting has its advantage in plant variety identification, and has transformed from indirect evidence into a mainstream method. A method for DNA fingerprinting called core loci combination was summarized in this study, which utilized a set of fixed core loci combination to identify different varieties. Since 2003, the project has been constantly improved and expanded. The first was to form combination of expanded loci by increasing the quantity of core loci to cope with increasing demand of variety identification of derived varieties. The second was to further decompose the core loci into group specific loci which played the function of fast and accurate clustering and species-specific loci which had stronger variety identification function. The third was to further propose the core loci combination of polyploidy crops based on the mature experience of diploid crop, which are diploidized single marker method and combination marker method. From the trend of future development, the rapid development of new technologies such as sequencing technology and gene editing technology will largely promote the improvement of DNA fingerprinting and improve the application effect in practice. This article has systematically elaborated the concept and characteristics of variety identification, summarized major types and identification methods of DNA fingerprinting, extracted technical methods for variety DNA fingerprinting and finally out-looked the future trend of DNA fingerprinting development. It is hoped that the study could provide guidances for the development of DNA fingerprinting standard of each crops, and the application of molecular technology in the structure and variety identification of DNA fingerprinting database.
The identification of plant varieties is of great significance for ensuring seed quality and food security in agricultural production. With the promulgation and implementation of seed law and supporting regulations, while the seed market is prospering, there are also ACTS such as infringement of brand name, producing and selling fake and inferior seeds, which affect the healthy and sustainable development of the seed industry (Wu et al., 2015). This is also the reason why the rapid and efficient variety identification method is urgently needed to escort the development of the seed industry. Compared with traditional identification techniques such as field planting identification and protein electrophoresis identification, DNA fingerprinting technology has unique advantages in variety identification. And SSR technology has the advantages of codominant inheritance, high polymorphism, simple data statistics, simple, fast process and easy standardization, and has been widely used in variety identification (Guichoux et al., 2011). China has been carried out the work of database construction on dozens of crops such as corn (Wang et al., 2017), rice (Xu et al., 2014), cotton (Kuang et al., 2015), wheat (Zheng et al., 2014), soybean (Gao et al., 2009), rapeseed (Ma et al., 2013) and Chinese cabbage (Zhang et al., 2013) and eatablished the identification standard based on SSR marker (Wang et al., 2015). SNP technology has the advantages of high flux and easy automation. High-flux SNP detection platforms have emerged, and chips for detecting thousands to tens of thousands of SNP sites have been introduced in corn (Unterseer et al., 2014), rice (Yu et al., 2014), wheat (Winfield et al., 2016) and other crops. InDel technology combines the advantages of SSR and SNP technology, and multiple InDel compound amplification system (Feng er al., 2017) has been developed, and the high-density chip of InDel has been successfully developed. In terms of variety area test (Wang et al., 2016), variety right protection (Xu, 2010), and seed quality supervision (Wu, 2015, China Seed Industry, 5:1-3), DNA fingerprint identification method has also changed from indirect evidence to mainstream method. However, in the field of variety identification, previous studies mainly focused on the technical level, involving few in detection schemes and without a systematic summary. This condition resulted in great differences in the actual application effect of molecular technology in different plants. But the widespread application of molecular identification has not been realized in many crops.
In recent years, many relevant seed industry documents released by the Ministry of Agriculture and the State Council, as well as the national long-term and medium-term scientific and technological development plan outline of the Ministry of Science and Technology, all involve the content of seed molecular detection and authentic DNA identification of varieties. In 2014, the General Principles of DNA Fingerprint Methods for Plant Variety Identification (hereinafter referred to as the General Principles) were promulgated and implemented. In 2015, the Ministry of Agriculture issued the "Construction of DNA Identification System for Crop Variety " program. The New Seed Law revised in 2016 takes DNA testing and other rapid test results as punishment basis. In 2017, the 13th Five-Year Key Research and Development Program of the Ministry of Science and Technology "Research and Application of Molecular Fingerprint Detection Technology for Seeds of Major Crops" was launched. The research and development of molecular technology such as SNP and the construction of shared database were carried out for maize, rice, wheat, soybean, cotton, rape and vegetable crops. This research was based on the repeated consideration and conclusion of the principles and strategies of plant variety identification in the process of the development of the General Principles, the construction of identification system and the revision of New Seed Law. This study systematically elaborated the concept and characteristics of variety identification, and the types and ideas of variety DNA fingerprint identification of varieties, summarized the different schemes of variety identification, and looked forward to the future development trend of the DNA fingerprint identification of varieties, with a view to playing a guiding role in the development of DNA fingerprint identification standards of crops and the application of molecular technology in the construction of DNA fingerprint database and the identification of varieties.
1 Concept and Characteristics of Plant Variety Identification
Identification refers to the identification of the true and false, merits and demerits of things. In the biological field, identification mainly involves species identification, species identification, individual identification and other types. Plant variety identification is a process of evaluating the plant population with certain morphological characteristics and production traits that have been artificially selected and bred. In terms of identification objects, variety identification is a type of identification between species identification and individual identification.
Variety identification and species identification are both based on plant population identification, and the main differences between the two are as follows: (1) Reproductive isolation is common among different species (De Queiroz, 2007), so the genetic differentiation among species is large and easy to distinguish, while there is no reproductive isolation between different varieties of the same species. Under the background of modern artificial breeding, germplasm resources in different areas have broken the geographical isolation, resulting in frequent combination and exchange of genome sequences and small genetic difference in the breeding varieties. (2) Species identification mainly makes use of gene (genome) sequence differences, namely haplotype DNA barcodes (Hebert et al., 2003; Moritz and Cicero, 2004), while variety identification mainly makes use of genotype differences, i.e. DNA fingerprint, which requires a combination of loci with high polymorphism below species level (Anastassopoulos, 2005).
Both variety identification and individual identification are identifications below species, and the main differences of the two are as follows: (1) Variety identification is an overall description of the homogeneous population of that variety rather than a separate description of each individual of the variety. Individual identification refers to the identification of each individual, which is applicable to the situation where there are great differences among different individuals within a group and it is meaningful to distinguish individuals. Individual identification is required less in plants, but more in humans and some animal species (Hill et al., 2009; Liu et al., 2013). (2) The same variety could be combined or propagated at many points for many years. For varieties of self-pollinated crops or inbred lines of cross-pollinated crops, the problem of residual heterozygous loci in varieties (lines) caused by insufficient self-crossing or mutation exists. For hybrid varieties, there are purity problems caused by inadequate isolation or emasculation measures during seed production, which may lead to some slight differences between seeds produced in different years and places (Yan et al., 2003; Xu et al., 2009). For individual identification, there is no variation in sampling of the same individual at different times. (3) The selection and breeding process of varieties is a process of artificial modification of materials by breeders, which can create a large number of varieties with highly similar genetic backgrounds. Therefore, variety identification needs to know the variation range of varieties in advance to serve as the determination threshold (Heckenberger et al., 2002). However, individual’s identification is faced with invariant individuals, and different individuals should have identifiable differences in the DNA sequences.
2 Identification Type and Idea of DNA Fingerprint of Varieties
2.1 Identification type
The identification of varieties DNA fingerprint is a process of comprehensive evaluation of varieties by using DNA fingerprint technology. The identification of varieties DNA fingerprint discussed in this study mainly involved four types: authenticity identification, derivative relationship identification, paternity identification, inbred line (or pure lines) tracing back. In addition to the above four types of genetically modified ingredients (Deng et al., 2011), male sterile identification (Zhang et al., 2005), functional gene identification (such as waxy gene, sweet genes, resistance genes, etc.) (Andersen and Lubberstedt, 2003), gender identification (Dong et al., 2006), reciprocal cross identification (Ge et al., 2013) belonged to the special type of cultivars. However, due to the identification of transgenic components, male sterility, functional genes and gender for specific traits, and the identification of positive and negative crossing for cytoplasmic differences, it was not included in the category of DNA fingerprint identification of varieties discussed in this study.
2.2 Identification idea
2.2.1 Authenticity identification
Authenticity identification is the identification of the true identity of varieties, focusing on whether there are significant differences between varieties, so the index used is the difference points. Authenticity identification process is a process of "seek differences", complete varieties of known standard DNA fingerprint database is the key to realize the identification, appraisal idea is as follows: the first step is to build a known breed standard DNA fingerprint database, which uses to distinguish the ability of a set of core loci (general dozens) of known breed standard sample build DNA fingerprint. The fingerprint database is characterized by high data quality and easy to share and could represent the "real fingerprint" of corresponding varieties. The second step is to compare varieties, it is using the same set of core sites for construction of DNA fingerprint check variety, according to comparing different testing requirements: (1) when used for screening variety label name corresponding standard fingerprint, the known breeds with the standard for check varieties corresponding varieties compare with the same DNA fingerprint libraries, tests confirmed for varieties of test sample name tagging is worthy of the name, namely the authenticity verification; (2) when the tested variety is anonymous or inconsistent with the label name, it shall be screened through the standard DNA fingerprint database of the known variety to determine the real variety name of the tested sample, namely, identity identification.
2.2.2 Identification of derived relations
The identification of variety derivation relations further answers whether those varieties that have been determined to be specific in the protection of variety rights are independent varieties or have derivation relations with the original varieties. Compared with authenticity identification, derivative relationship identification focuses more on the degree of genetic background similarity among varieties, so the index used is genetic similarity. The identification process of derivative relationship is a process of "finding similarity", and determining the appropriate threshold is the key to realize identification. The identification idea is as follows: the first step is to determine the appropriate threshold of derivative varieties. Using a set of core loci uniformly selected from the whole genome, a DNA fingerprint database is constructed for a set of representative varieties of this species. According to the genetic similarity among varieties, and combining the phenotype and genealogy information, the appropriate genetic similarity threshold with a high degree of expected coincidence is determined. Among them, the determination of derived varieties and the same varieties should be further combined with the phenotypic difference. The second step is to identify the derived varieties, that is, using the same set of core loci to construct the DNA fingerprint of the two varieties to be tested, calculate the genetic similarity of the two varieties, use the above threshold, and determine whether the two varieties are derived varieties by combining the phenotypic differences. Based on the studies of Noli et al. (2013) and Heckenberger et al. (2002), the relationship diagram of independent varieties, derived varieties and original varieties was formed (Figure 1).
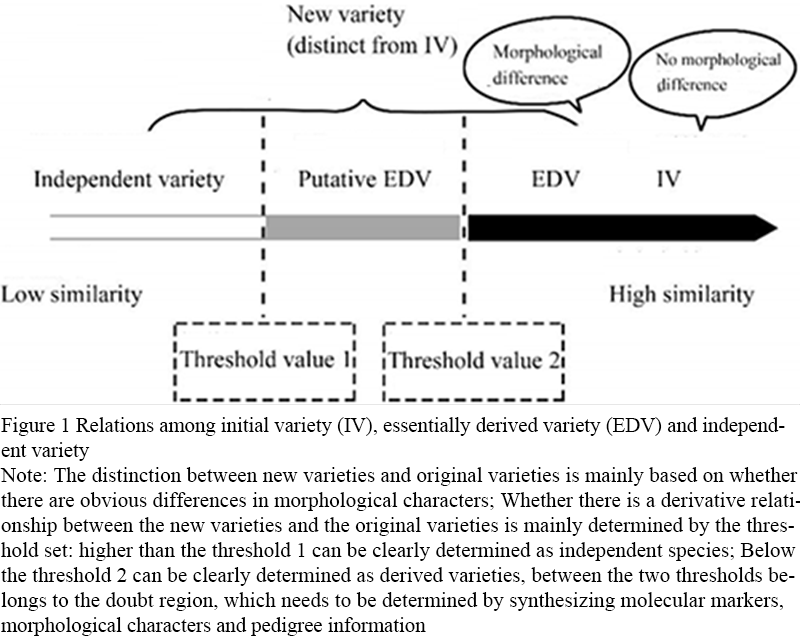 Figure 1 Relations among initial variety (IV), essentially derived variety (EDV) and independent variety Note: The distinction between new varieties and original varieties is mainly based on whether there are obvious differences in morphological characters; Whether there is a derivative relationship between the new varieties and the original varieties is mainly determined by the threshold set: higher than the threshold 1 can be clearly determined as independent species; Below the threshold 2 can be clearly determined as derived varieties, between the two thresholds belongs to the doubt region, which needs to be determined by synthesizing molecular markers, morphological characters and pedigree information |
2.2.3 Parental test
Paternity test is the identification of the parent inbred line in the previous generation with retroactive hybrid combinations, which could be divided into triplets, dizygotic and parents suspected (Li et al., 2005): (1) the so-called triplets’ referees to the provision of a hybrid and its known parent 1 and the hypothesis parent 2 to determine whether the hypothesis parent 2 is its true parent. (2) the so-called dizygotic referees to the provision of a hybrid and its hypothetical parent 1 to determine whether the hypothetical parent 1 is its true parent. (3) When both parents were doubtful, it means to provide the hybrid and its hypothesized parents 1 and 2 to determine whether "hypothesized parents 1 and hypothesized parents 2" are its true parents. Of course, in cotton, rapeseed and other crops, there is still the phenomenon of reuse of F2 generation seeds. Tracing F1 generation hybrid seeds according to F2 generation seeds is also a special paternity test (Chen et al., 2014).
The thinking of paternity has four: (1) using SSR, SNP codominant markers, using female parent organization such as the seeds on the skin, the skin, such as the skin of corn, rice husk of rice, beans, melons of pod shells, such as direct access to maternal genotype, and combined with hybrid genotypes, according to Mendelian inheritance law, estimate the male parent genotype (Zhao Jiu however, etc., 2004). The idea is to obtain the genotypes of both hybrids and their parents in the case that the parents are unknown and only the hybrid seeds are present. (2) We using tissues with different proportions of maternal and paternal components, such as endosperm, and combining with codominant markers, to decompose the parental genotypes (Ge et al., 2013). The ratio of maternal and paternal components in endosperm of cereal crops was 2:1 (Zhang G.S., and Zhao H.Y., 1990, Biological Bulletin, 12: 17-19; Mo, 1995), using the co-dominant marker of heterozygous symmetric amplification for typing, the amplification product quantity of the two alleles of maternal and paternal should be 2:1, so the allele with the ratio of 2 comes from the maternal and the allele with the ratio of 1 comes from the paternal. This idea can be applied to the seeds with large endosperm and easy exfoliation, such as the seeds of monocotyledonous plants such as corn and wheat. (3) According to the genotype of the hybrids, the triad or diad algorithm (Yang Q.G., 1998, Chinese Journal of Forensic Science, 13(2): 90-92) was used to screen the possible parent inbred lines from the known inbred line pool, or the two-parent doubting algorithm (Lu et al., 2001) was used to screen the possible parent combinations from the known inbred line pool. This idea is applicable to a wide range, but need to have database support. (4) Using maternal genetic components such as chloroplast, the possible maternal inbred lines of hybrids were speculated. This idea is limited by the low degree of sequence variation in cytoplasmic components such as chloroplasts and mitochondria (Palmer and Herbon, 1988; Heinze, 2007), It can only be used as an aid to paternity testing.
2.2.4 Ancestry of inbred lines (or pure lines)
Inbred (or purebred) ancestry is the identification of possible ancestors in inbred (or purebred) varieties. According to the actual needs of identification, it can be divided into two situations. One is to identify which existing germplasm resources have been used by breeders in the selection and breeding of new inbred lines or pure lines as the parents of the selected lines, generally not more than ten generations back. The second is to identify inbred lines of possible ancient ancestors, up to the generation of thousands of years. There are two approaches for this kind of identification: (1) using the differences in cytoplasmic genomes such as chloroplast and mitochondria to trace the maternal ancestry. The cytoplasmic components, such as chloroplast and mitochondria, are inherited from the maternal line, without the occurrence of genome recombination and exchange, and the genome sequence is conservative, which can be traced back to the maternal ancestors of a distant generation (Liu et al., 2008, Henan Agricultural Science, 37(7): 5-9; Dong et al., 2012). The disadvantage of this scheme is that it can only be traced back to the mother, not the father. (2) nuclear genome differences are used to trace ancestors. By finding regions with low recombination rate (or no recombination) in nuclear genome (Gao et al., 2005), and further screening regions with high mutation rate in these regions, these regions can play the progenitor role similar to chloroplast and mitochondrial genome, and can be traced back to both parents.
3 DNA fingerprint identification scheme of varieties
3.1 Establishment the method of core loci set
The first scheme of variety identification is to use a set of fixed core loci to identify different varieties. Since the core locus grouping method was first proposed in 2003, it has played an important guiding role in the molecular identification of crop varieties (Wang et al., 2003). Dozens of crops have screened the core primer combination of this crop successively (Li et al., 2010; Wang et al., 2011; Bai et al., 2012; Sui et al., 2014), and applied in the construction of standard DNA fingerprint database and the identification of varieties. Core loci was preferable when varieties DNA fingerprint of a set of site, has the characteristics of high polymorphism, good repeatability, uniform distribution, unified for varieties of DNA molecular marker data acquisition and cultivars of site, comprehensive UPOV organize to set up the BMT molecular test guide and China promulgated the "general plant variety identification of DNA fingerprint method" relevant content, the selected indicators core of loci including high polymorphism, easy to statistical data, good repeatability, good compatibility, chromosome distribution of different platform clear, uniform distribution in the genome, avoid choose zero allelic variation, etc. Different crop core loci combination to determine the main basis different genera (species) of plants varieties quantity, difference situation between varieties, the chromosome number, genome size and level of polymorphic loci, different markers, and give attention to two or more things site flux characteristics of test platform, test the speed and cost, and can distinguish the plant genera (species) more than 95% of the known species, the core site portfolio site number between tens to hundreds of commonly.
3.2 The improvement and development of core loci set
At present, the core loci combination, which developed and applied on crops is obtained from the selection of large-scale loci uniformly selected from the whole genome, but the value of different markers is different, and the impact on the analysis results is also different. Most plant species have a large number of varieties. Due to ecological adaptation, breeding habits and geographical isolation, they have formed their own unique variety groups. If ignoring the variety group and facing a large number of varieties directly, it will increase the difficulty of variety identification. Taking corn as an example, the number of maize varieties and combinations has reached tens of thousands, and the inbred lines of corn have formed different heterosis groups such as sipingtou, luda honggu, red, Lanka and P groups, with obvious differences among groups (Liu et al., 2012). In view of this, if the identification of varieties is carried out in two steps, a small number of markers will be used to determine the group of varieties to which the varieties belong, and then the further differentiation of different varieties within the group will be conducive to the accurate and rapid identification of varieties, especially when the number of varieties is large. Accordingly, the core site is divided into two levels, level 1 is the core of the differentiate varieties of site, will breed because of geographical isolation, breeding habits, ecological form of varieties such as clear as a number of varieties, can achieve the goal of tag insertion loss variation could be big pieces, it is possible that low restructuring and the area of high mutation, based on the three generations of sequencing technology development. The second level is the core locus to identify varieties, and to identify small differences in the same species, such as SSR, SNP, InDel and other small sequence variations, so as to distinguish different varieties within the variety group. The selection of variety identification markers depends on the degree of variation within each variety group. Conversely, the lower the degree of variation, the more varieties there are and the more markers are needed. Therefore, when calculating the polymorphism of markers, it is necessary to calculate the markers with high polymorphism in each variety group for each variety group, and find the markers with high polymorphism in each variety group as the preferred markers for the species. Taking the identification of maize inbred lines as an example, it can be grouped according to the situation of heterosis group. First, a set of specific core loci of the group can be found to classify the varieties into different heterosis groups, and then the varieties within the group can be further distinguished, and the specific loci of the variety can be found to distinguish different varieties. The species-specific loci and the species-specific loci of the variety group combine to form a set of core loci combinations. This is a further improvement on the original core site grouping method. The species-specific loci have the function of rapid and accurate classification, while the species-specific loci have the function of strong variety identification.
With the emergence of a large number of essentially derived varieties (EDV), which puts forward higher requirements for the variety identification, the number of choice points and the ability to distinguish the available points need to be optimized urgently. Therefore, on the basis of the original core points, the number of additional points is added to form the extended point combination. Extend the basic principles of site selection and core sites are the same, but more, and more focus on chromosome evenly distributed, when used with a core site should be able to distinguish between the plant genera (species) more than 99% of the known species, number of sites around several thousand to tens of thousands of (according to different crop varieties of genome size and diversity). The following factors should be considered in the selection of extension site combination: (1) the significance of different variation sites is different, and the selection of intra-gene or inter-gene, and the selection of genes affecting the function or not; (2) the selection of loci was based on physical location or genetic linkage map; (3) different types of variation loci, such as SSR, SNP and InDel, have different meanings.
The principle of core locus grouping method: the identification loci include two categories of core loci and extension loci, in which the core loci are divided into two levels of grouping loci and variety differentiation loci, and the number of loci is within the range of 1-10 and 10-100, respectively. Extension sites can be divided into several levels, and the number of sites increases gradually (Figure 2A). In the face of a large number of varieties, the varieties were first divided into several groups by the grouping sites in the core loci, and then the different varieties in the group were identified by the differentiation loci of the varieties in the core loci. For the varieties still unable to be distinguished, the extended core loci were used for identification (Figure 2B).
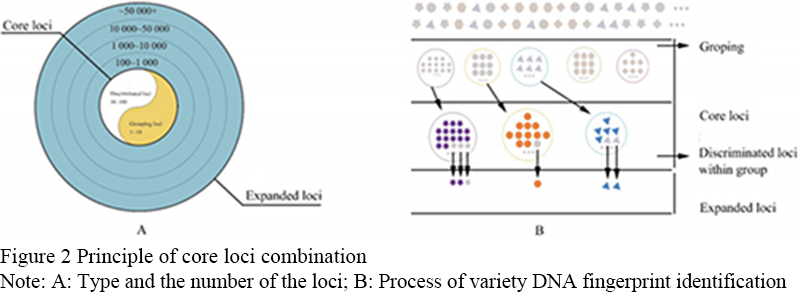 Figure 2 Principle of core loci combination Note: A: Type and the number of the loci; B: Process of variety DNA fingerprint identification |
3.3 effects of crop ploidy on core loci combination selection
The core locus grouping method has been widely used in the construction of DNA fingerprint database and variety identification of diploid plant varieties such as maize (Lu et al., 2014; Wang et al., 2017). Due to the complexity of genome brought by ploidy in polyploid plants (Yang ji, 2001), it is difficult to select core loci, which has an impact on the application of core locus grouping method in the identification of polyploid plant varieties. Therefore, on the basis of learning from the mature experience of diploid plants, two possible strategies for the combination selection of core loci suitable for the characteristics of polyploid crops were proposed, namely the diploid single labeling method and the combination labeling method.
Diploid single-marker method: studies on the evolution of polyploid genomes have proved that the long-term evolutionary process eventually leads to the diploidy of polyploid plants (Wolfe, 2001). Therefore, for most polyploid plant species, the screening of diploid polymorphic sites is highly feasible in practice. The strategy includes three situations: first, polymorphic markers are designed for genomic regions that are missing from the corresponding chromosome subgroups, and their expression is similar to that of diploid crops (Figure 3A; Figure 3B). The second is to select different sequences between different chromosomal subgroups, which can be distinguished, and only one chromosomal subgroup has sequence polymorphism. Polymorphic markers are designed on this region to only count the information of the chromosomal subgroup with polymorphism and filter out the information of other non-polymorphic chromosomal subgroups (Figure 3C). The third is to choose between different chromosome subgroup of the sequence is different, can distinguish, and in different sequence polymorphism of chromosome subgroup is different, in this design on the area of polymorphic markers, according to the sequence polymorphism is able to distinguish between different chromosome subgroup, and further statistical each chromosome polymorphism of subgroups (Figure 3D). The figure shows the site selection strategy of the diploid single-marker method by taking the heterotetraploid species as an example. The first four pictures show the situation that diploidy can be achieved. The marker type can be SSR, InDel or SNP, and it can be classified by electrophoresis or sequencing (Figure 3A; Figure 3B; Figure 3C; Figure 3D); The last picture shows the situation where diploidy is not possible. Because the sequence of different chromosome subgroups cannot be distinguished, the diploidy markers cannot be designed for these regions (Figure 3E).
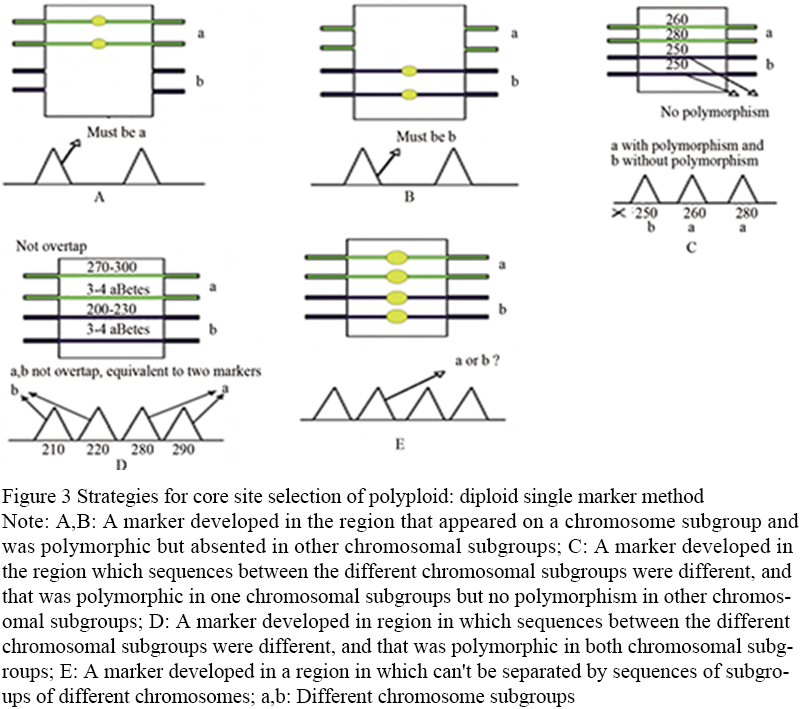 Figure 3 Strategies for core site selection of polyploid: diploid single marker method Note: A,B: A marker developed in the region that appeared on a chromosome subgroup and was polymorphic but absented in other chromosomal subgroups; C: A marker developed in the region which sequences between the different chromosomal subgroups were different, and that was polymorphic in one chromosomal subgroups but no polymorphism in other chromosomal subgroups; D: A marker developed in region in which sequences between the different chromosomal subgroups were different, and that was polymorphic in both chromosomal subgroups; E: A marker developed in a region in which can't be separated by sequences of subgroups of different chromosomes; a,b: Different chromosome subgroups |
Combinatorial labeling means that since it is often difficult to achieve the goal of distinguishing different subgroups and having polymorphism within subgroups by only one marker, the second strategy is to select at least two closely adjacent markers to form combinatorial markers. Combinatorial markers include two types, one plays the role of distinguishing different chromosomal subsets, and the other plays the role of providing further sequence polymorphism within the subsets. These markers were shown to be co-isolated without recombinant exchange. Since only the overall combination of these markers could play a role, only the sequencing platformcould be used for typing. Figure of allotetraploid species, for example, shows the site selection strategy of combinatorial labeling (Figure 4), six times, eight times and other polyploid species can be used in a similar strategy: choose closely adjacent two polymorphic loci form loci combination, one of the loci (generally SNPS or InDel) to distinguish the various subgroups, another one (generally SSR) to distinguish polymorphism within the subgroups, sequencing of loci combination area, using the sequencing result to part.
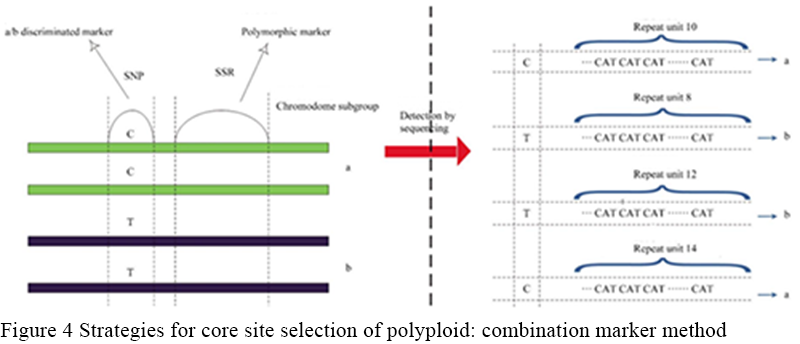 Figure 4 Strategies for core site selection of polyploid: combination marker method |
4 Relationship between morphological identification and DNA fingerprint identification
Similar to DNA fingerprint identification by selecting a set of core loci combination, in terms of the protection of plant variety rights, a set of variety identification system based on morphological traits was also established - DUS test (Wang et al., 2009). A set of suitable morphological traits were selected for phenotypic data collection, and the differences among the identified varieties were compared by database comparison or by paired and side-planted comparison. Since the goal of both identification systems is to identify varieties, it is inevitable to face the problem of how to combine the results of phenotype identification and DNA fingerprint identification. In the framework of International Union for The Protection of New Varieties of Plants (UPOV), put forward three possible ways of combination: one is to use morphological chain or molecular markers in the gene, that is function marker to predict the morphological characters, this way is difficult, only in some resistance character, male sterility character researches, but still can't be totally corresponded. Second is to build correlation between morphological characters difference and molecular marker character difference, this way is the focus of current research. It has been explored in the corn (Babic et al., 2016), barley (Jones et al., 2013), and other crops. The preliminary results show that there is a certain correlation between them, but there is a non-linear correlation. Based on the existing research results, an application model of combining morphological traits with molecular markers was established. Thirdly, DNA fingerprints are used as an independent identification system different from morphological traits to establish their own identification rules. This method has been adopted in human identification (Hill et al., 2009). Although there is no consensus on plant species identification, it has made a useful attempt to establish the molecular identification systems of SSR and SNP on maize, and it shows that there is a high correlation between the two systems (Rousselle et al., 2015).
5 Future Prospects
Development of sequencing technology, chip technology, gene editing technology, nucleic acid thermostatic amplification technology and other technologies (Thompson and Milos, 2011; Sander and Joung, 2014; Zhao et al., 2016), will greatly promote the improvement of DNA fingerprint identification scheme and improve its application effect in practice: (1) Combining the third-generation sequencing technology and the more perfect InDel development algorithm (Lv et al., 2016), the mining of the insertion deletion sites of large fragments is faster and more efficient, and such sites are more likely to have better clustering effect, thus making the development of clustering markers in the core locus grouping method easier. With the improvement of second-generation sequencing technology, it is not only more accurate in marker typing (Churbanov et al., 2012; Li et al., 2017), and it is possible to achieve the target detected by SNP, SSR, InDel and other types of markers on a sequencing detection platform at the same time, so that the core locus grouping method is no longer limited to the detection platform in marker selection, but is possible to freely select various types of sequence variation regions. (2) With the development and improvement of high-density DNA chip technology, more loci can be detected with less cost, so that the number of extended loci can be distributed more evenly and densely on the whole gene, to eliminate the deviation caused by fewer loci, and to evaluate the genetic similarity of derived varieties more accurately. (3) With the development of gene editing techniques and mature, is expected to achieve the design of various molecular sequence tags fast into the varieties, and even the DNA sequence of a can be decoded as video (Shipman et al., 2017), like product label before delivery, as a specific tag of cultivars, bred variety rights protection in agriculture in the enterprise play an important role. (4) with the constant temperature amplification technology of nucleic acid, the target nucleic acid sequence amplification can be achieved at a constant temperature without temperature cycle, which simplifies the requirements of the instrument and makes it easier to achieve the “rapid and simple” identification goal.
Author’s Contributions
WFG and THL are the conceivers of the review, responsible for data collection, analysis and the writing of the first draft of the paper. ZJR conducted overall guidance and check on the content of the paper; YHM, ZH, LYD, KM and ZLK participated in the data collection and analysis of the paper, and put forward suggestions for revision of the first draft of the paper; HYX is responsible for the paper chart drawing; DMQ is responsible for English translation and polishing of abstracts. All authors read and agree on the final text.
Acknowledgement
This study was funded by the national key research and development program (2017YFD0102001), the national science and technology support program (2015BAD02B02) and the Beijing academy of agricultural and forestry sciences’ college-level science and technology innovation team construction project (JNKYT201603).
Anastassopoulos E., 2005, DNA fingerprinting in plants: principles, methods and applications, Economic Botany, 14 (14):129-131
Andersen J.R., and Lubberstedt T., 2003, Functional markers in plants, Trends Plant Sci., 8(11): 554-560
https://doi.org/10.1016/j.tplants.2003.09.010
PMid:14607101
Babic V., Nikolic A., Andjelkovic V., Kovacevic D., Filipovic M., Vasic V., and Mladenovic-Drinic S., 2016, AUPOV morphological versus molecular markers for maize inbred lines variability determination, Chilean Journal of Agricultural Research, 76(4): 417-426
https://doi.org/10.4067/S0718-58392016000400004
Bai J., Nie Y.C., Lin Z.X., Guo X.P., Zhang X.L., Wang B., and Liu C.X., 2012, Screening and evaluation of SSR core primers for identification of cotton hybrids, Mianhua Xuebao (Cotton Science), 24(3): 207-214
Chen Q.Q., Zhan X.J., Lan J.Y., Huang Y., Fu J.P., and Zhang Z.Y., 2014, Research advance of utilization and development on hybrid cotton F2, Xiandai Nongye Keji (Modern Agricultural Science and Technology), (23): 65-67
Churbanov A., Ryan R., Hasan N., Bailey D., Chen H., Milligan B., and Houde P., 2012, High SSR: high-throughput SSR characterization and locus development from next-gene sequencing data, Bioinformatics, 28(21): 2797-2803
https://doi.org/10.1093/bioinformatics/bts524
PMid:22954626 PMCid:PMC4439519
De Queiroz K., 2007, Species concepts and species delimitation, Syst. Biol., 56(6): 879-886
https://doi.org/10.1080/10635150701701083
PMid:18027281
Deng H.C., Yin C.C., Liu G.Z., Lin J.R., and Deng P.J., 2011, Progress in nucleic acid detection techniques for genetically modified organisms, Zhongguo Shengwu Gongcheng Zazhi (China Biotechnology), 31(1): 86-95
Dong L.N., Su X., Sun K., Zhang J.Q., Zhang H., and Chen W.,2006, Applications of DNA molecular markers on sex identification of dioecious plants, Guangxi Zhiwu (Guihaia), 26(1): 63-68
Dong W., Liu J., Yu J., Wang L., and Zhou S., 2012, Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding, PLoS One, 7(4): e35071
https://doi.org/10.1371/journal.pone.0035071
PMid:22511980 PMCid:PMC3325284
Feng B., Xu L.W., Wang F.G., Xue N.N., Liu W.B., Yi H.M., Tian H.L., Lv Y.D., Zhao H., Jin S.Q., Zhang L.K., Yu R.H., and Zhao J.R., 2017, Establishment of multiplex PCR system with 20 pairs of InDel markers in maize, Zuowu Xuebao (Acta Agronomica Sinica), 43(8): 1139-1148
https://doi.org/10.3724/SP.J.1006.2017.01139
Gao L., Mu X.Q., Lin Y., Li Y.G., and Cheng Z.K., 2005, Meiotic recombination hotspots in Eukaryotes, Yichuan (Hereditas), 27(4): 641-650
Gao Y.L., Zhu R.S., Liu C.Y., Li W.F., Jiang H.W., Li C.D., Yao B.C., Hu G.H., and Chen Q.S., 2009, Establishment of molecular ID in soybean varieties in Heilongjiang, China, Zuowu Xuebao (Acta Agronomica Sinica), 35(2): 211-218
https://doi.org/10.3724/SP.J.1006.2009.00211
Ge M., Jiang L., Zhang X.L., Zhao H., and Zhang T.F., 2013, The principle of distinguishing maize hybrids of direct and reciprocal crosses using insertion/deletion (InDel) markers and its application, Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 11(1): 37-47
Guichoux E., Lagache L., Wagner S., Chaumeil P., Léger P., Lepais O., Lepoittevin C., Malausa T., Revardel E., Salin F., and Petit R.J., 2011, Current trends in microsatellite genotyping, Molecular Ecology Resources, 11(4): 591-611
https://doi.org/10.1111/j.1755-0998.2011.03014.x
PMid:21565126
Hebert P.D., Cywinska A., Ball S.L., and Dewaard J.R., 2003, Biological identifications through DNA barcodes, Proc. Biol. Sci., 270(1512): 313-321
https://doi.org/10.1098/rspb.2002.2218
PMid:12614582 PMCid:PMC1691236
Heckenberger M., Bohn M., Ziegle J.S., Joe L.K., Hauser J.D., Hutton M., and Melchinger A.E., 2002, Variation of DNA fingerprints among accessions within maize inbred lines and implications for identification of essentially derived varieties. 玉. Genetic and technical sources of variation in SSR data, Mol. Breed., 10(4): 181-191
https://doi.org/10.1023/A:1020539330957
Heinze B., 2007, A database of PCR primers for the chloroplast genomes of higher plants, Plant Methods, 3(1): 4Hill C.R., Butler J.M., and Vallone P.M., 2009, A 26plex autosomal STR assay to aid human identity testing, J. Forensic. Sci., 54(5): 1008-1015
https://doi.org/10.1111/j.1556-4029.2009.01110.x
PMid:19627417
Jones H., Norris C., Smith D., Cockram J., Lee D., O'Sullivan D.M., and Mackay I., 2013, Evaluation of the use of high-density SNP genotyping to implement UPOV model 2 for DUS testing in barley, Theor. Appl. Genet., 126(4): 901-911
https://doi.org/10.1007/s00122-012-2024-2
PMid:23232576
Kuang M., Wang Y.Q., Zhou D.Y., Fang D., Ma L., and Yang W.H., 2015, Construction of SSR fingerprinting database of standard varieties on cotton in DUS testing, Mianhua Xuebao (Cotton Science), 27(1): 46-52
Li H.B., Yang J., Lv Z.W., Yi B., Wen J., Fu T.D., Tu J.X., Ma C.Z., and Shen J.X., 2010, Screening of Brassica napus core SSR primers, Zhongguo Youliao Zuowu Xuebao (Chinese Journal of Oil Crop Sciences), 32(3): 329-336
Li L., Fang Z., Zhou J., Chen H., Hu Z., Gao L., Chen L., Ren S., Ma H., Lu L., Zhang W., and Peng H., 2017, An accurate and efficient method for large-scale SSR genotyping and applications, Nucleic Acids Res., 45(10): e88
https://doi.org/10.1093/nar/gkx093
PMid:28184437 PMCid:PMC5449614
Li X.H., Li X.H., Gao W.W., Tian Q.Z., Li M.S., Ma F.M., and Zhang S.H., 2005, Establishment of DNA fingerprinting database of maize hybrids and its application in parentage identification, Zuowu Xuebao (Acta Agronomica Sinica), 31(3): 386-391
Liu G., Liu C.S., Li N., Han X., and Wang J., 2013, Establishment of DNA database for individual identification andparentageanalysis using microsatellites in Chinese hosteinbulls (Bos taurus), Nongye Shengwu Jishu Xuebao (Journal of Agricultural Biotechnology), 21(9): 1085-1092
Liu Z.Z., Wu X., Liu H.L., Li Y.X., Li Q.C., Wang F.G., Shi Y.S., Song Y.C., Song W.B., Zhao J.R., Lai J.S., Li Y., and Wang T.Y., 2012, Genetic diversity and population structure of important Chinese maize inbred lines revealed by 40 core simple sequence repeats (SSRs), Zhongguo Nongye Kexue (Scientia Agricultura Sinica), 45(11): 2107-2138
Lu H.L., Yang Q.E., and Hou Y.P., 2001, The calculation of PI value in case of paternity testing of alleged parents, Zhongguo Fayixue Zazhi (Chinese Journal of Forensic Medicine), 16(4): 210-213
Lu X.Z., Ni J.L., Li L., Wang X.F., Ma H., Zhang X.J., and Yang J.B., 2014, Construction of rice variety identity using SSR fingerprint and commodity information, Zuowu Xuebao (Acta Agronomica Sinica), 40 (5): 823-829
https://doi.org/10.3724/SP.J.1006.2014.00823
Lv Y., Liu Y., and Zhao H., 2016, M InDel: a high-throughput and efficient pipeline for genome-wide InDel marker development, BMC Genomics, 17(1): 290
https://doi.org/10.1186/s12864-016-2614-5
PMid:27079510 PMCid:PMC4832496
Ma L., Liu H.Z., Lu X.Z., Ni J.L., Zhang X.J., and Yang J.B.,2013, Molecular identity of 130 Brassica napus varieties, Zhongguo Youliao Zuowu Xuebao (Chinese Journal of Oil Crop Sciences), 35(3): 231-239
Mo H.D., 1995, Genetic research of endosperm-quality traits in cereals, Zhongguo Nongye Kexue (Scientia Agricultura Sinica), 28(2): 1-7
Moritz C., and Cicero C., 2004, DNA barcoding: promise and pitfalls, PLoS Biol., 2(10): e354
https://doi.org/10.1371/journal.pbio.0020354
PMid:15486587 PMCid:PMC519004
Noli E., Teriaca M.S., and Conti S., 2013, Criteria for the definition of similarity thresholds for identifying essentially derived varieties, Plant Breeding, 132(6): 525-531
https://doi.org/10.1111/pbr.12109
Palmer J.D., and Herbon L.A., 1988, Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence, J. Mol. Evol., 28(1-2): 87-97
https://doi.org/10.1007/BF02143500
PMid:3148746
Rousselle Y., Jones E., Charcosset A., Moreau P., Robbins K., Stich B., Knaak C., Flament P., Karaman Z., Martinant J.P., Fourneau M., Taillardath A., Romestant M., Tabel C., Bertran J., Ranc N., Lespinasse D., Blanchard P., Kahler A., Chen J., Kahler J., Dobrin S., Warner T., Ferris R., and Smith S., 2015, Study on essential derivation in maize: III. Selection and evaluation of a panel of single nucleotide polymorphism loci for use in european and north American germplasm, Crop Sci., 55(3): 1170-1180
https://doi.org/10.2135/cropsci2014.09.0627
Sander J.D., and Joung J.K., 2014, CRISPR-Cas systems for editing, regulating and targeting genomes, Nat. Biotechnol., 32(4): 347-355
https://doi.org/10.1038/nbt.2842
PMid:24584096 PMCid:PMC4022601
Shipman S.L., Nivala J., Macklis J.D., and Church G.M., 2017, CRISPR-Cas encoding of a digital movie into the genomes of a population of living bacteria, Nature, 547 (7663): 345-349
https://doi.org/10.1038/nature23017
PMid:28700573 PMCid:PMC5842791
Sui G.L., Yu S.C., Yang J.X., Wang W.H., Su T.B., Zhang F.L., Yu Y.J., Zhang D.S., and Zhao Y.Y., 2014, Validation of acore set of microsatellite markers and its application for varieties identification in Chinese cabbage, Yuanyi Xuebao (Acta Horticulturae Sinica), 41(10): 2021-2034
Thompson J.F., and Milos P.M., 2011, The properties and applications of single-molecule DNA sequencing, Genome Biol.,12(2): 217
Unterseer S., Bauer E., Haberer G., Seidel M., Knaak C., Ouzunova M., Meitinger T., Strom T.M., Fries R., Pausch H., Bertani C., Davassi A., Mayer K.F.X., and Schön C.C., 2014, A powerful tool for genome analysis in maize: development and evaluation of the high density 600 k SNP genotyping array, BMC Genomics, 15(1): 823
https://doi.org/10.1186/1471-2164-15-823
PMid:25266061 PMCid:PMC4192734
Wang F.G., Tian H.L., Zhao J.R., Yi H.M., Wang L., and Song W., 2011, Development and characterization of a core set of SSR markers for fingerprinting analysis of Chinese maize varieties, Maydica, 56(1): 7-17
Wang F.G., Yang Y., Yi H.M., Zhao J.R., Ren J., Wang L., Ge J.R., Jiang B., Zhang X.C., Tian H.L., and Hou Z.H., 2017, Construction of an SSR-based standard fingerprint database for corn variety authorized in China, Zhongguo Nongye Kexue (Scientia Agricultura Sinica), 50 (1): 1-14
Wang F.G., Yi H.M., Zhao J.R., Sun S.X., Yang G.H., Ren J., and Wang L., 2016, The application of DNA fingerprint technology in maize varieties' authenticity and consistency identification in maize regional test, Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 14(2): 456-461
Wang F.G., Zhao J.R., Guo J.L., She H.D., and Chen G., 2003, Comparison of three DNA fingerprint analyzing methods for maize cultivars' identification, Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 1(5): 655-661
Wang F.G., Zhao J.R., Tian H.L., Yang Y., and Yi H.M., 2015, The progress of the crop varieties DNA fingerprint database construction, Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 13(9): 2118-2126
Wang F.G., Zhao J.R., Yi H.M., and Song W., 2009, Application of DNA fingerprint technology on maize varieties protection, Yumi Kexue (Journal of Maize Sciences), 17(6): 136-139
Winfield M.O., Allen A.M., Burridge A.J., Barker G.L.A., Benbow H.R., Wilkinson P.A., Coghill J., Waterfall C., Davassi A., Scopes G., Pirani A., Webster T., Brew F., Bloor C., King J., West C., Griffiths S., King I., Bentley A.R., and Edwards K.J., 2016, High-density SNP genotyping array forhexaploid wheat and its secondary and tertiary gene pool, Plant Biotechnol. J., 14(5): 1195-1206
https://doi.org/10.1111/pbi.12485
PMid:26466852 PMCid:PMC4950041
Wolfe K., 2001, Yesterday's polyploids and the mystery of diploidization, Nat. Rev. Genet., 2(5): 333-341
https://doi.org/10.1038/35072009
PMid:11331899
Xu L., Liu Y.H., Shi Y.S., Song Y.C., Wang T.Y., and Li Y.,2009, Genetic uniformity and genetic variation of maize inbred lines with same name but different origin revealed by SSR markers, Zhiwu Yichuan Ziyuan Xuebao (Journal of Plant Genetic Resources), 10(1): 32-36
Xu Y.B., ed., 2010, Molecular plant breeding, Science Press, Beijing, China, pp.492-538
https://doi.org/10.1079/9781845933920.0000
Yan Z., Chang R.Z., Guan R.X., Liu Z.X., and Qiu L.J., 2003, Analysis of similarity and difference of soybean varieties Mancangjins from various regions by using agronomic traits and SSR markers, Zhiwu Yichuan Ziyuan Xuebao (Journal of Plant Genetic Resources), 4(2): 128-133
Yang J., 2001, The formation and evolution of polyploid genomes in plants, Zhiwu Fenlei Xuebao (Acta Phytotaxonomica Sinica), 39(4): 357-371
Yu H., Xie W., Li J., Zhou F., and Zhang Q., 2014, A whole genome SNP array (RICE6K) for genomic breeding in rice, Plant Biotechnol. J., 12(1): 28-37
https://doi.org/10.1111/pbi.12113
PMid:24034357
Zhang W., Cui J.Z., Yu S.C., and Li L., 2013, Construction of SSR fingerprint database of Chinese cabbage varieties (Brassica campestris L. ssp. Pekinensis), Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 11(6): 843-857
Zhang Z.X., Fang M.J., Du H.W., Deng L.R., and Zheng Y.L.,2005, The rapid discrimination based on PCR on cytoplasmic types of male sterile line of maize (Zea mays L.), Zuowu Xuebao (Acta Agronomica Sinica), 31 (10): 1386-1388
Zhao J.R., Liu L.Z., Wang F.G., Guo J.L., and Wang Y.D., 2004, Identification authenticity of female parent of F1 hybrids through SSR profiling of pericarp tissue in hybrid, Yumi Kexue (Journal of Maize Sciences), 12(3): 6-8
Zhao X., and Hou X.H., 2016, Analysis of the research trend of isothermal nucleic acid amplification technology, Jiyinzuxue Yu Yingyong Shengwuxue (Genomics and Applied Biology), 35(3): 656-663
Zheng Y.S., Zhang H., Wang D.J., Sun J.M., Wang X.M., Duan L.L., Li H., Wang W., and Li R.Y., 2014, Development of a wheat variety identification system based on fluorescently labeled SSR markers, Zhongguo Nongye Kexue (Scientia Agricultura Sinica), 47(19): 3725-3735



. PDF(714KB)
. FPDF(win)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Fengge Wang
. Hongli Tian
. Hongmei Yi
. Han Zhao
. Yongxue Huo
. Meng Kuang
. Like Zhang
. Yuanda Lv
. Manqing Ding
. Jiuran Zhao
Related articles
. Plant
. Cultivar identification
. DNA fingerprinting
. Core loci
. Extended loci
Tools
. Email to a friend
. Post a comment