Research Report
Development of Polymorphic SSR Markers in Vallisneria Based on RAD-seq 
2 Water Environment & Ecology Engineering Research Center of Shanghai Institution of Higher Education, Shanghai 201306, China
 Author
Author  Correspondence author
Correspondence author
Molecular Plant Breeding, 2021, Vol. 12, No. 30 doi: 10.5376/mpb.2021.12.0030
Received: 24 Sep., 2021 Accepted: 30 Sep., 2021 Published: 09 Oct., 2021
Tan M., Shi Y.X., Zheng H.S., Shao L., and He P.M., 2021, Development of polymorphic SSR markers in Vallisneria based on RAD-seq, Molecular Plant Breeding, 12(30): 1-7 (doi:10.5376/mpb.2021.12.0030)
Vallisneria belongs to Hydrocharitaceae, which is a pioneer species of ecological restoration in waters. In order to carry out a more comprehensive SSR analysis ofVallisneria, we used the restriction-site associated DNA sequencing (RAD-seq) technology to analyze Simple Sequence Repeats (SSR) information of Vallisneria, SSR markers and primer of Vallisneria were developed based on RAD-seq data.Among which ,366 simple sequence repeats (SSR) loci were detected. Primers of 355 loci were designed successfully, and the two base repeat type SSR loci were the largest (59.56%). Finally, we confirmed 23 pairs of polymorphic SSR primers after the screening and validation. The analysis results by Genepop software showed that the mean number of alleles of the 23 loci is 3.26, and these loci do not link to each other (P<0.01). Four loci deviate from HWE (P<0.01) and the number of heterozygous roughly the same as that of homozygotes (observed heterozygosity mean of 0.460), inbreeding coefficient is high (mean of 0.880). Which could be caused by take sample too closely in the same sampling, andVallisneria have a combination of generative propagation and clone.
Vallisneria belongs to the Hydrocharitaceae. It is a perennial stemless submerged herb with stolon and linear or ribbon-shaped leaves. It grows in creeks, rivers and other environments (Chen et al., 2006) and is widely distributed all over the world (Lowden, 1982). It is a constructive species and an important part of freshwater ecosystems. Vallisneria has a strong ability to adsorb suspended pollutants and has the ability to remove nitrogen and phosphorus. It can effectively alleviate the degree of eutrophication of water bodies, making it a pioneer species in water purification and water ecological restoration, and is widely favored (Irfanullah and Moss, 2004).
Simple Sequence Repeat (SSR) is microsatellite (Litt et al., 1989), refers to a set of DNA formed by multiple repetitions of a unit composed of 1 to 6 nucleotides in the genome, which are distributed in various regions of the genome. The length of each region is usually within the range of 200 bp. SSR markers have the advantages of rapid and convenient detection, strong reliability, high experimental reproducibility and high polymorphism (Zhang, 2007, Journal of Anhui Agricultural Sciences, (4): 972-975), which are widely used in the construction of gene maps, the comparison and identification of kinship relationships, the location of functional genes and QTLs, and the use of population genetic diversity, genetic structure and origin (Schlötterer and Tautz, 1992; Lv and Guo, 2018).
At present, the research on Vallisneria mainly focuses on taxonomy (Les et al., 2008), reproductive ecology (Xiong and Li, 2000; Chen et al., 2006), water purification function (Bolpagni et al., 2015), physiological effects on environmental factors, etc. (Iriel et al., 2015). These studies have conducted in-depth explorations on the physiological characteristics and ecological functions of Vallisneria, but there are few advances in molecular biology analysis such as genetic diversity. At present, Chen et al. (2006) have used allozymes and developed 11 pairs of microsatellite markers to study the genetic diversity and genetic structure of V. spinulosa in the middle and lower reaches of the Yangtze River. Wang et al. (2010) used 8 pairs of ISSR microsatellite primers, and Wang et al. (2011) used 22 pairs of SSR primers to explore the genetic variation and genetic system of the Vallisneria race in Dianshan Lake, Niushan Lake and other regions. Wang (2017) used nSSR and cpSSR molecular marker methods to explore the genetic characteristics and genetic systems of six species of Vallisneria in the vicinity of Dagang River, but there is no comprehensive SSR information analysis of Vallisneria. Since there is no reference genome for Vallisneria, this study would use restriction-site associated DNA sequencing (restriction-site associated DNA, RAD-seq) (Miller et al., 2007) to carry out detailed analysis on the SSR information of Vallisneria, and develop its polymorphic molecular markers accordingly to provide references for the screening and application of polymorphic SSR markers.
1 Results and Analysis
1.1 Simplified Genome Quality Inspection of Vallisneria
The results of the simplified genome of Vallisneria (Table 1), the average Q20 (sequencing base quality value was 1% of the probability of misjudgment during sequencing, the percentage of bases with an accuracy of 99%) was 98.6%; the average Q30 (sequencing bases quality value was 0.1% of the probability of misjudgment during sequencing, the percentage of bases with an accuracy of 99.9%) was 95.33%, and the average GC percentage was about 42%. The difference before and after filtering was very small. Q20 and Q30 were both at a high level, which proved that the amount of sample information was sufficient, the sequencing quality was up to standard, the GC division was normal, and the sequencing information was reliable (Table 1).
|
Table 1 Summary of genomic sequences generated by RAD-seq |

1.2 The frequency and distribution of SSR sites in the simplified genome of Vallisneria
Using MISA software, a total of 366 SSR sequences were obtained, but no multiple repeated motif positions were found. All sequences were located in single repeated motif positions. The length of the obtained SSR was 16~62 bp, and the average length was 30 bp. The largest number was the site with a length of 16 bp, 62 bp and 30 bp were the least; the length division of each repetitive sequence showed great differences, and the number of repetitions of the motif related to the number of nucleotides also had certain differences (Table 2). Dinucleotide SSR was basically repeated 6 to 7 times (approximately 72%); trinucleotide SSR was basically repeated 5 to 6 times (approximately 83%); tetranucleotide SSR was mainly repeated 4 times (The ratio was about 47%). Overall, among all motif repeats, six repeats were the most, with a total of 134 (36.61%). Followed by five repetitions, a total of 95 (25.96%), and only 8 (2.18%) more than 15 repetitions.
|
Table 2 Distribution of SSR in the reduced-representation genome |

Among the 366 SSR sites mentioned above, 335 SSR sites (approximately 92%) were successfully designed using Primer 3.0 software.The repetitive motif changes were based on between 1 and 6 bases: the number of two-base repetitive motifs was the largest, accounting for 59.56% of the total. The number of repetitions was 6~30, and the average length of the repetitive sequence was 16 bp. The types of repetitive sequence were mainly TA, AT, CT, AG, TC, which accounted for 15.27%, 14.48%, 7.37%, 6.83%, 4.10% of the total number of SSR sequences, respectively. There were 126 three-base repetitive motifs, accounting for about 47% of the total. The number of repetitions was 5~20, and the average length was 18 bp. The types of repetitive sequences were mainly GAA, TTA, TTC, TCT, the frequencies were 2.19%, 2.19%, 2.19%, 1.91%, respectively. There were 15 four-base repeat motifs, the number of repetitions was 5~15, and the average length of the repetitive sequence was 26 bp. The main repeat type was TACA, and the frequency was 0.82%.
1.3 SSR verification
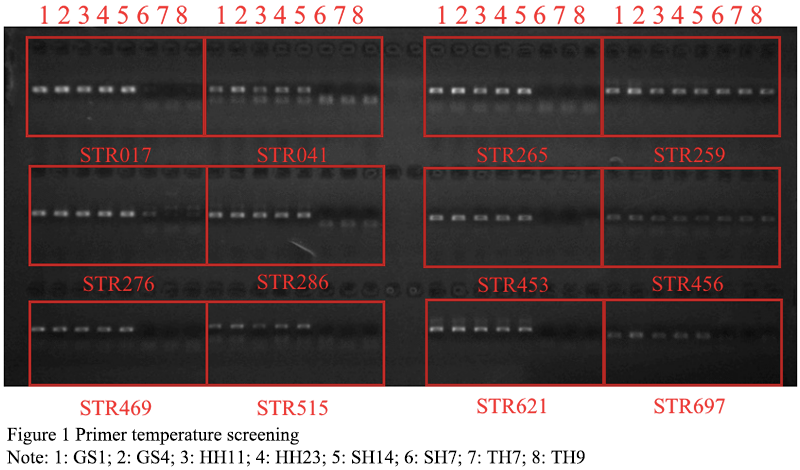
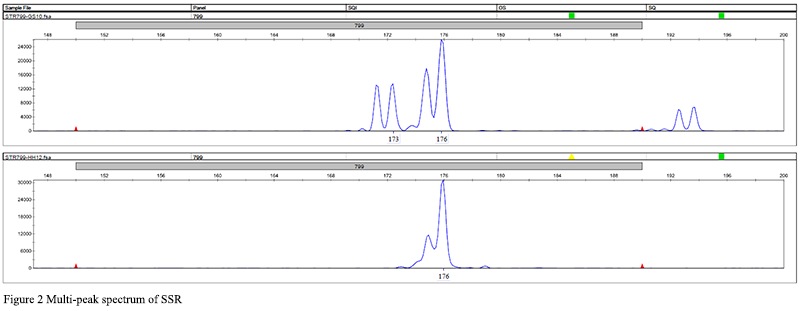
180 pairs of primers were selected from 335 pairs of primers for polymorphism screening and detection. The obtained reaction product was separated by electrophoresis using a 1.5% agarose gel, and analyzed by the imaging system to successfully amplify a single stable target band primer. If there was a clear band, it could be determined that the primer was effective, and follow-up operations could be carried out (Figure 1). In the research graph obtained with Gene Mapper, the relative fragment size of the highest peak of the sample was obtained. If the peak type and fragment length in each amplification result did not match, it proved that the primer had the attribute of polymorphism. A total of 33 sites with preliminary polymorphisms were obtained through polymorphism screening, and 25 sites were further selected for peak types to be easily screened for verification of a large population sample size (Figure 2). After typing 25 sites in 120 samples, the peak patterns of STR3116 and STR11173 were different from those in the screening experiment, and the alleles could not be accurately and stably determined, so they were excluded. Finally, it was clarified that 23 of the 180 pairs (approximately 13%) SSR primers showed polymorphic properties (Table 3). Among the 23 pairs of SSRs, the two-base and three-base repetitive motifs were 17 and 6, respectively, which accounted for 16% and 9% of the number of motif positions to be selected respectively. The fragment size of the PCR amplified product with 23 pairs of primers was between 125 and 303 bp, and the number of alleles observed was between 2 and 7, and the average number of alleles per locus was 3.26 (Table 3).
|
Figure 1 Primer temperature screening Note: 1: GS1; 2: GS4; 3: HH11; 4: HH23; 5: SH14; 6: SH7; 7: TH7; 8: TH9 |
|
Figure 2 Multi-peak spectrum of SSR |
|
Table 3 Characterization of the 23 SSR loci in Vallisneria Note: NA: Number of alleles per locus |
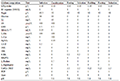
Using Genepop detection, it was found that 23 pairs of polymorphic SSR primers amplified a total of 120 samples from 6 Vallisneriapopulations to obtain a total of 75 alleles. The average observed heterozygosity Ho of the 6 Vallisneria populations was 0.460, and the expected heterozygosity He was 0.342. Only STR96350, STR40400, STR104040, and STR33880 had a slight deviation from Hardy Weinberg equilibrium (P>0.05). The coefficient of inbreeding Fis was between -1 and 1, with an average value of 0.880.
2 Discussion
In this study, the simplified genome sequence of Vallisneria was successfully obtained by using the RAD sequencing technology of next-generation sequencing. At the same time, the SSR information of Vallisneria was studied based on this, and 335 pairs of SSR primers were developed for it, and finally 23 pairs of polymorphic SSR sequences were screened out of the selected 180 pairs, which had no single base repeat motif sequence. The development efficiency of using simplified genome sequencing technology was much higher than that of Chen et al. (2006) using magnetic bead enrichment method to develop microsatellite primers from the genome of V. spinulosaofficinalis. A total of 29 pairs of microsatellite primers were obtained from the enrichment, and 26 pairs successfully amplified the target fragments, of which 11 pairs were polymorphic amplified. The Genepop test showed that the mean value of its alleles was 3.26, and the polymorphism was high; the 4 loci deviated from Hardy Weinberg equilibrium, which may be due to the two ways of asexual and sexual reproduction in the genus Vallisneria (Xiong and Li, 2000), the asexual reproductive method of Vallisneria could quickly occupy a wider habitat in order to absorb nutrients (Madsen, 1991). The ratio of homozygote to heterozygote was equal (the average observed heterozygosity was 0.460), which may be because the genus Valeriana had two reproduction methods at the same time. The coefficient of inbreeding was relatively high, with a mean Fis value of 0.880, that is, 88% of the genetic variation of the Vallisneria population existed among the populations, and only 12% existed within the populations. This result was contrary to the previous research results of Chen et al. (2008) and Wang (2018), this may be because the six populations sequenced this time were geographically far apart. During the sampling process, the same population was collected in a concentrated manner, so it was greatly affected by asexual reproduction.
In this study, RAD-seq technology was used for simplified genome sequencing of Vallisneria for the first time. A total of 259 984 093 high-quality clean reads and a total of 366 SSR loci were obtained, mainly based on dinucleotides and trinucleotides repetitive type. 335 pairs of primers were designed, and 23 pairs of SSR sites with polymorphism were finally screened out, and the development efficiency was high. This research provided abundant and reliable data for the future study of Vallisneria using RAD-seq technology, and the research results would be helpful to the research of Vallisneria breeding, molecular marker development, phylogenetic research, genetic protection, etc. The TIANGEN DP305 kit was used to extract the leaf genomic DNA from the selected materials of sorrel grass (Guo et al., 2019), and 1.5% agarose gel electrophoresis and ultraviolet spectrophotometer were used to define the attributes of the DNA to ensure that the quality of the obtained genomic DNA can meet the requirements. Library conditions.
3 Materials and Methods
3.1 Experimental Materials
The materials used in this study were taken from the "Taihe Water" breeding base (Shanghai Fengjing), Taihu Lake (Suzhou), Honghu Lake (Jingzhou), Shanghai Luchao Port, Hongze Lake (Huai'an), Huangshan Xin'an River (Table 4), 30 adult individuals of Vallisneriawere randomly selected at each sampling point. The TIANGEN DP305 kit was used to extract the leaf genomic DNA from the selected materials of Vallisneria (Guo et al., 2019), and 1.5% agarose gel electrophoresis and ultraviolet spectrophotometer were used to define the attributes of the DNA to ensure that the quality of the obtained genomic DNA can meet the conditions for building a database.
|
Table 4 Location of the sampling sites |
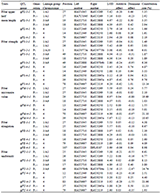
3.2 Simplify genome sequencing
The 8 genomic DNAs involved in the four related groups were fused in equal amounts, and the database was constructed and sequenced by Shanghai Maipu Biotech. The operation process was as follows: restriction endonuclease digests genomic DNA, plus P1 connector (P1 connector contained primer sequence required for PCR amplification, Illumina sequencing primer binding site sequence and short tag sequence to distinguish different samples); mixed the samples with the corresponding P1 linker and physically changed them into a sequence of 300~500 bp; added P2 connector; added enriched RAD-tags in PCR, constructed pair-end files, and carried out high-throughput sequencing in the Illumina MiSeq PE3150 system; screened, detected, and evaluated the initial information (raw data) to obtain high-quality sequencing data (clean reads). Use trimmomatic software for filtering, the filtering steps were as follows: eliminated linker sequences in reads, eliminated reads that had no inserted fragments due to linker interconnection and other factors; modified bases with low quality tails (quality results less than 20), and eliminated sequences containing bases with a quality value of less than 10; eliminated reads with an N ratio of more than 10%; eliminated adapter and quality-modified sequences that are less than 75 bp in length.
3.3 SSR site analysis and SSR primer design
MISA (http://pgrc.ikpgatersleben.de/misa) software was used to estimate the qualified SSR. At the same time, Primer 3 (Li and Durbin, 2010) software was used to develop SSR primers for better SSR sites.
3.4 SSR site screening
Site polymorphism screening mainly included singleness and polymorphism screening. The singleness was determined by agarose gel after PCR to determine whether there is a product and whether the product is single. Polymorphism screening was used to determine whether the peak shape was qualified and polymorphic by capillary electrophoresis after fluorescent primer PCR. Screening experiments were carried out using 8 samples from 6 populations (GS8, GS18, SH11, HH28, TMTH25, HA4, HS14, HS17), and 180 sites were screened for polymorphism in 7 times, and PCR amplification was carried out by tailing Primer, typing primer Common-famF (FAM-AGTCACGACGTTGTAAAACGAC). The amplification system and procedures were as follows: in a 20 μL PCR reaction mechanism, 1 μL of 30 ng/μL DNA template, 1.0 μL typing primer, 0.2 μL each of 10 μmol/L forward primer, and 1 μL reverse primer, Tag polymerase 10 μL, ddH2O 7 μL. PCR amplification was performed on the Mastercycler pro PCR machine: treatment at 94°C for 5 min; treatment at 94°C for 30 s and annealing treatment at 55°C~57°C for half a minute; delay at 72°C for 30 s, and these three steps need to be carried out in sequence 35 times; add 15 min at 72°C. Then, the PCR products were electrophoresed with 1% agarose and stained with ethidium bromide (EB), observed and photographed in the gel imaging system.
Authors’ contributions
TM and SYX designed and carried out the study. TM completed the data analysis and drafted the manuscript. HPM conceived of the project. HPM, ZHS and SL directed data analysis, draft and revision. All authors read and approved the final manuscript.
Acknowledgments
This study was supported by "Thirteenth Five-Year" Water Project: Suzhou Regional Water Quality Improvement and Water Ecological Safety Assurance Technology and Comprehensive Demonstration Project (2017ZX07205).
Bolpagni R., Laini A., Soana E., Tomaselli M., and Nascimbene J., 2015, Growth performance of Vallisneria spiralis under oligotrophic conditions supports its potential invasiveness in mid-elevation freshwaters, Weed Research, 55(2): 185-194
https://doi.org/10.1111/wre.12128
Chen K.N., Lan C.J., Shi L.X., Chen W.M., Xu H., and Bao X.M., 2006, Reproductive ecology of Vallisneria natans, Zhiwu Shengtai Xuebao (Acta Phytoecologica Sinica), (3): 487-495
Chen L., Les D.H., Xu L.M., Yao X.H., Kang M., and Huang H.W., 2006, Isolation and characterization of a set of microsatellite loci in the submerged macrophyte, Vallisneria spinulosa Yan (Hydrocharitaceae), Molecular Ecology Resources, 6(4): 1243-1245
https://doi.org/10.1111/j.1471-8286.2006.01503.x
Chen L., Ye Q.G., Pan L.Z., Xu L.M., and Huang H.W., 2008, Vallisneria species in lakes of the middle-lower reaches of the Yangtze River of China, Zhiwu Shengtai Xuebao (Acta Phytoecologica Sinica), (1): 106-113
Guo C.Y., Li S.X., Su G.C., Saren Q.M.G., and Hasi A., 2019, Functional analysis of CmERFII-9 gene in melon fruit ripening, Jiyinzuxue Yu Yingyong Shengwuxue (Genomics and Applied Biology), 38(10): 4610-4616
Irfanullah H.M., and Moss B., 2004, Factors influencing the return of submerged plants to a clear-water, shallow temperate lake, Aquatic Botany, 80(3): 177-191
https://doi.org/10.1016/j.aquabot.2004.07.010
Iriel A., Lagorio M.G., and Cirelli A.F., 2015, Biosorption of arsenic from groundwater using Vallisneria gigantea plants. Kinetics, equilibrium and photophysical considerations, Chemosphere, 138: 383-389
https://doi.org/10.1016/j.chemosphere.2015.06.053
PMid:26143400
Les D.H., Jacobs S.W.L., Tippery N.P., Chen L., Moody M.L., and Wilstermann-Hildebrand M., 2008, Systematics of Vallisneria (Hydrocharitaceae), Systematic Botany, 33(1): 49-65
https://doi.org/10.1600/036364408783887483
Li H., and Durbin R., 2010, Fast and accurate long-read alignment with Burrows-Wheeler transform, Bioinformatics, 26(5): 589-595
https://doi.org/10.1093/bioinformatics/btp698
PMid:20080505 PMCid:PMC2828108
Litt M., and Luty J.A., 1989, A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene, Am. J. Hum. Genet., 44(3): 397-401
Lowden R.M., 1982, An approach to the taxonomy of Vallisneria L. (Hydrocharitaceae), Aquatic Botany, 13(3): 269-298
https://doi.org/10.1016/0304-3770(82)90064-X
Lv Z.H., and Guo H.C., 2018, Comparison of genetic diversity analysis by RSAP, SSR and SRAP markers in potato, Jiyinzuxue Yu Yingyong Shengwuxue (Genomics and Applied Biology), 37(6): 2544-2550
Madsen J.D., 1991, Resource allocation at the individual plant level, Aquatic Botany, 41(1): 67-86
https://doi.org/10.1016/0304-3770(91)90039-8
Miller M.R., Dunham J.P., Amores A., Cresko W.A., and Johnson E.A., 2007, Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers, Genome Research, 17(2): 240-248
https://doi.org/10.1101/gr.5681207
Schlötterer C., and Tautz D, 1992, Slippage synthesis of simple sequence DNA, Nucleic Acids Research, 20(2): 211-215
https://doi.org/10.1093/nar/20.2.211
Wang B., Liao H., Zhao Y., Li W., and Song Z.P., 2011, Microsatellite loci in Vallisneria natans (Hydrocharitaceae) and cross-reactivity with V. spinulosa and V. denseserrulata, American Journal of Botany, 98(3): e44-e47
https://doi.org/10.3732/ajb.1000441
Wang B., Song Z.P., Liu G.H., Lu F., and Li W., 2010, Comparison of the extent of genetic variation of Vallisneria natans and its sympatric congener V. spinulosa in lakes of the middle-lower reaches of the Yangtze River, Aquatic Botany, 92(4): 233-238
https://doi.org/10.1016/j.aquabot.2009.12.006
Wang D.B., 2017, Analyis of the influence of Dagang River mater quality change on genetic diversity and genetic structure of Vallisneria natans, Thesis for M.S., School of Life Sciences, Nanjing University, Supervisor: Wang Z.S., pp.6
Xiong B.H., and Li W., 2000, Ecological studies on Vallisneria L. in China, Wuhan Zhiwuxue Yanjiu (Journal of Wuhan Botanical Research), 18(6): 500-508



. PDF(925KB)
. HTML
Associated material
. Readers' comments
Other articles by authors
. Meng Tan
. Yuxin Shi
. Haisu Zheng
. Liu Shao
. Peimin He
Related articles
. Vallisneria
. RAD-seq
. SSR molecular marker
. Reduced-representation genome sequencing
Tools
. Email to a friend
. Post a comment