Association mapping of six agronomic traits on chromosome 4A of wheat (Triticum aestivum L.) 
2.National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, 430070, P.R. China
 Author
Author  Correspondence author
Correspondence author
Molecular Plant Breeding, 2010, Vol. 1, No. 5 doi: 10.5376/mpb.2010.01.0005
Received: 26 Jul., 2010 Accepted: 20 Oct., 2010 Published: 30 Oct., 2010
Liu et al., 2010, Association mapping of six agronomic traits on chromosome 4A of wheat (Triticum aestivum L.), Molecular Plant Breeding Vol.1 No.5 (doi: 10.5376/mpb.2010.01.0005)
Association mapping is a powerful approach to identify associations between traits of interest and genetic markers. In this study, 103 wheat germplasm accessions from China were genotyped using 76 SSR markers and 40 EST-SSR markers. The phenotyping of plant height, spike length, spikelets per spike, spikelets density, grains per spike and thousand-kernel weight were carried out in three locations for three years. Six subpopulations were identified among these accessions by population structure analysis based on 49 SSR and 40 EST-SSR markers. Linkage disequilibrium (LD) on chromosome 4A extended up to ~3 cM with r2=0.054. Based on the mixed linear model considering population structure and relative kinship, a total of 10 SSR markers (p<0.01) on chromosome 4A were significantly associated with six agronomic traits, and six of them were associated with multiple traits. Some of the associated markers were in agreement with previous quantitative trait loci (QTL) analysis. This study demonstrated that association mapping can be successfully applied in wheat breeding context for detection of marker-traits associations. Furthermore, association mapping can enhance previous QTL information and provide additional QTL information for marker-assisted selection.
Background
For molecular breeders the goal of plant genetics research is to identify genes or genomic regions that are responsible for the phenotypes (Weigel and Nordborg, 2005), and most traits of agricultural or evolutionary importance are controlled by multiple quantitative trait loci (QTLs). In the past several decades, QTLs studies in wheat and many other crop species were mainly carried out by linkage analysis in F2-, DH (double haploid)- or RIL (recombinant inbred lines)- derived mapping populations based on genetic recombination. However, with the development of molecular biology and biometrics, association mapping as a new and powerful tool has been demonstrated that it could complement and enhance previous QTL information identified by linkage analyses for marker-assisted selection in wheat (Breseghello and Sorrells, 2006). Association mapping is a method to detect correlations between genotypes and phenotypes in a collection of germplasm based on linkage disequilibrium (LD). A major advantage of this approach over classical linkage analysis in breeding germplasm is that it does not require the time-consuming and expensive generation of specific genetic populations (Flint-Garcia et al. 2005). Furthermore, this approach can detect larger number of alleles and increase mapping resolution (Yu and Buckler 2006).
Association mapping has been first successfully used for genetic studies on some complex human diseases, and it has also been used successfully in various plant species to identify markers associated with a variety of phenotypes in recent years. Significant associations have been identified between SNPs (single nucleotide polymorphisms), RFLP (restriction fragment length polymorphism), AFLP (amplified fragment length polymorphism), SSR (single sequence repeat) markers and varied agronomical or morphological traits in maize (Thornsberry et al. 2001; Beló et al. 2008; Harjes et al. 2008), rice (Agrama et al., 2007;Wen et al. 2009), barley (Kraakman et al. 2006) and soybean (Jun et al. 2007). In hexaploid wheat, Breseghello and Sorrels (2006) firstly conducted association test between kernel traits and SSR markers in 95 elite wheat germplasms. Thereafter, different types of molecular markers were identified to be associated with some disease resistance traits such as stem rust resistance, leaf rust resistance, yellow rust resistance, powdery mildew resistance, stagonospora nodorum blotch resistance, russian wheat aphid resistance, fusarium head blight resistance and yield in wheat (Rhone et al. 2007; Crossa et al. 2007; Tommasini et al. 2007; Peng et al. 2008; Zwart et al. 2008). All these studies showed that association mapping is a very effective tool for QTL research in wheat.
One major obstacle in applying association mapping to crop species is that the complex breeding histories of many important crops have created complex population structures within the germplasms (Flint-Garcia et al. 2003). Spurious associations can arise in the presence of population structure and unequal distribution of alleles within subpopulations (Lander and Schork 1994). Recently, population structure can be successfully detected based on the Bayesian model method using unlinked markers distributing through whole genome (Pritchard et al. 2000a, b). The mixed linear model considering population structure and relative kinship (can be detected by marker-based estimation of the probability of identity by descent between individuals) in association mapping were employed to control both false positive and false-negative rates (Yu et al. 2006). Recently, this approach has been successfully practiced for association mapping in wheat (Breseghello and Sorrels, 2006; Yao et al. 2009). Furthermore, in order to control spurious associations, rare alleles (with frequency<5%) in the population can be treated as missing data for population structure, linkage disequilibrium analysis and association mapping (Breseghello and Sorrells, 2006).
In previous studies, a number of QTLs for agronomically important traits have been identified on chromosome 4A in wheat by linkage mapping approach, including plant height, heading date, grain yield, tiller number per plant, spike number per unit area, spike length, spikelet number, grain number, compactness and grain weight (Araki et al., 1999; Börner et al., 2002; Li et al. 2002; Jantasuriyarat et al. 2004; Kirigwi et al. 2007; Kumar et al. 2007). However, these QTLs were generally localized in larger intervals by linkage mapping. With the development of wheat SSR maps (Röder et al. 1998; Pestsova et al. 2000; Gupta et al. 2002; Somers et al. 2004), QTL mapping on chromosome 4A using marker-traits association mapping is feasible.
The objectives of this research are: (1) to estimate population structure among a collection of 103 wheat germplasm accessions; (2) to estimate the extent of LD and LD blocks; (3) to analyze the association of SSR markers on chromosome 4A with six agronomic traits. The results of this study will help wheat breeders to identify specific targets for wheat genetic improvement.
1 Results
1.1 Marker polymorphism
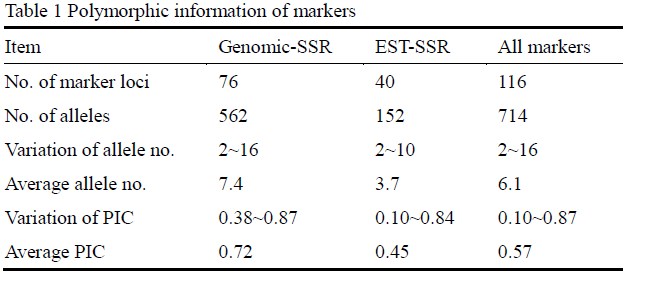
A total of 76 SSR and 40 EST-SSR markers were scored across 103 wheat accessions. Eighty-nine unlinked markers including 49 SSR and 40 EST-SSR markers were used for population structure assessment, and 31 SSR markers on 4A were used for association analysis. A total of 714 alleles were amplified at 116 markers among the 103 wheat accessions, and the number of alleles per locus ranged from 2 to 16 with an average of 6.1. Besides, the average PIC value was 0.57 with a range of 0.10~0.87 (Table 1). The results also showed that the SSR markers had more alleles and higher PIC than these EST-SSR markers (Table 1). After taking out the rare alleles (about 5.8%), the effective allele numbers for all of the loci varied from 2 to 7 with an average of 3.80.
|
Table 1 Polymorphic information of markers
|
1.2 Population structure
The model-based analysis with Structure identified an optimal number of sub-populations when K was set at 6, because the likelihood peaked at K = 6 in the range of two to twelve subpopulations. The number of these 103 wheat accessions assigned to each of the six inferred clusters ranged from 8 to 28. FST values between all groups were significant (P<0.001) and ranged from 0.36 to 0.71, suggesting a real difference among these clusters and supporting the existence of genetic structure.
1.3 Linkage disequilibrium
The background LD (unlinked LD) in the genome created by population structure was used to determine specific critical value of LD on 4A chromosome markers. The unlinked r2 were obtained by analysis of a total of 89 unlinked markers. The unlinked r2 values ranged from 0.000 to 0.299 for all unlinked loci pairs with an average of 0.021. The 95th percentile of the distribution of unlinked r2 estimates was used as a population-specific threshold for this parameter as an evidence of linkage (Breseghello and Sorrels, 2006), and the critical value is 0.054. The syntenic r2 were obtained by analysis of 31 SSR markers on chromosome 4A, which covered the whole chromosome. The syntenic r2 values ranged from 0.000 to 0.388 with an average of 0.031, significantly higher than the average of unlinked r2. Decay scatterplots of the LD values based on the syntenic r2 value for 103 accessions are shown in Figure 1. The extent of LD on chromosome 4A was ~3 cM with the critical value r2 =0.054, and about 89% of LD pairs were detected with r2 <0.054.
|
Figure 1 Scatterplot of the LD (r2) against genetic distance (cM) between all loci pairs on chromosome 4A. Horizontal straight lines indicate the 95th percentile of the distribution of unlinked r2
|
.png)
LD block was referred to a chromosomal region where all pairs of adjacent loci were in LD (Stich et al. 2005). Three LD blocks were detected on 4A. The first one was observed in the centromeric region, including barc206, barc138 and wmc420 within 3.5 cM (positions 3.6~7.1 cM,Figure 2). The second one was in the long arm, including gwm637, wmc468, wmc707 and wmc161 within 9.5 cM (positions 36.5~46 cM,Figure 2). The last one was in the bottom of 4A long arm, including wmc313, wmc497 and wmc219 within 4.7 cM (positions 82.9~87.6 cM, Figure 2).
|
Figure 2 Consensus map of loci tested on chromosome 4A (Somers et al., 2004)
|
.png)
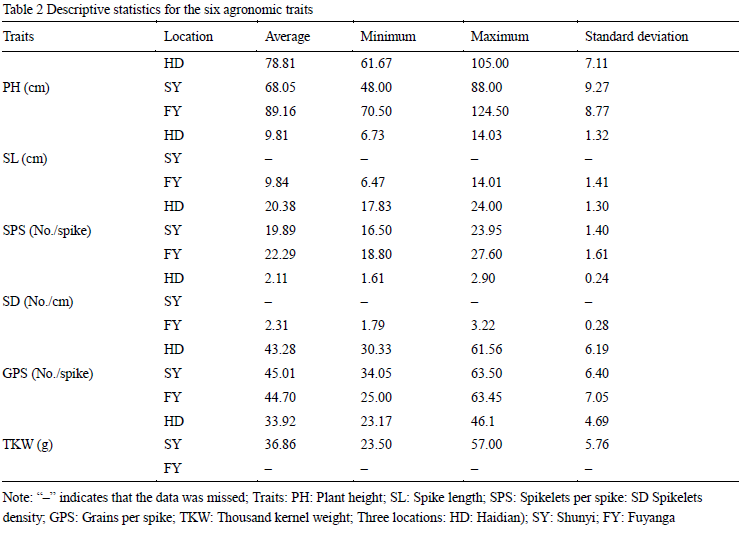
1.4 Phenotypic data
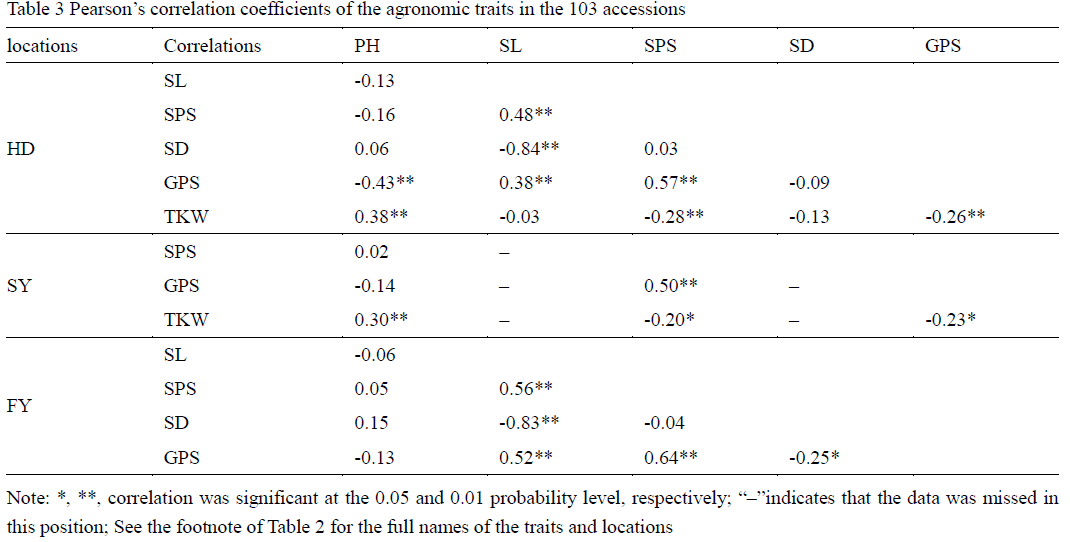
The phenotypic data used in this study was based on the mean values across three years in three environments (HD, SY and FY). PH, SPS and GPS were evaluated in HD, SY and FY. SL and SD were evaluated in HD and FY, whereas TKW was evaluated in HD and SY (Table 2). PH, SL, SPS, GPS and SD were the largest in FY among three locations, probably because of more favorable weather conditions in FY. The results of the analysis of variance of six traits among three locations indicated that environment had a large effect on PH, SPS, TKW and SD, whereas it had a small effect on SL and GPS. Pearson correlation coefficients of the agronomic traits in the 103 accessions were also evaluated (Table 3). In general, TKW was positively correlated to SL, SPS, SD and GPS, but negatively correlated to PH. In addition, positive correlations were also detected among SL, SPS and GPS. PH was negatively correlated to SL, SPS and GPS, negatively correlated to HD and TKW, which showed that PH can have on many other agronomic characteristics.
|
Table 2 Descriptive statistics for the six agronomic traits
|
|
Table 3 Pearson’s correlation coefficients of the agronomic traits in the 103 accessions
|
1.5 Association mapping
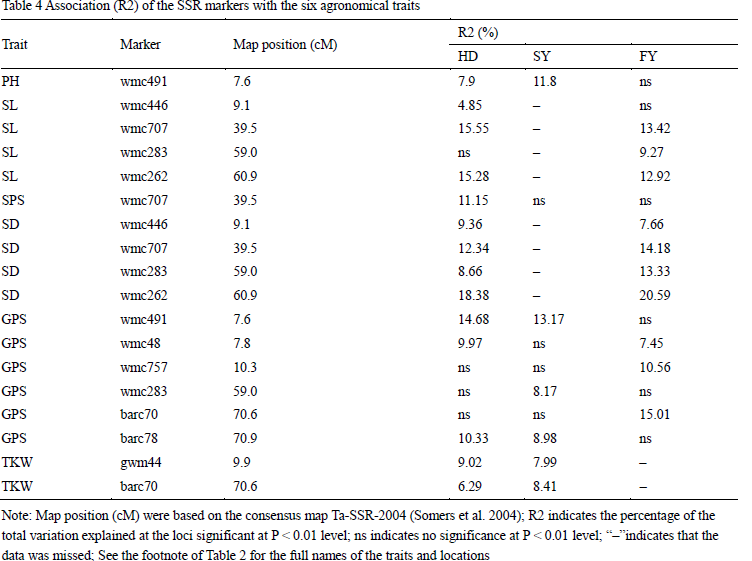
In the present study, we used the association mapping approach to searching QTLs for the six agronomic traits in different locations (Table 4). A total of 10 SSR markers were identified to be associated with the six traits at the 0.01 probability level, and each QTL explained 4.85% to 20.59% of phenotypic variation. There were one, two, four and six SSR markers showed significant correlation with PH and SPS, TKW, SL , SD, and GPS, respectively. Wmc491 was associated with PH and GPS both in HD and SY. Wmc262, wmc707, wmc283 and wmc446 was significantly correlated to SL and SD simultaneously in HD and FY, and explained similar phenotypic variation. Wmc707 was only associated with SPS in one location (HD). Wmc48, barc78 and wmc491 showed significant correlation with GPS in two locations, respectively. Barc70, wmc757 and wmc283 were detected to be significantly associated with GPS in one location, respectively. Gwm44 and bac70 were also detected to be significantly associated with TKW in two locations. In conclusion, most marker-trait associations can be detected in two locations, but no one marker showed significant association with traits in three locations, this might be caused by missing data (only three traits were investigated in all of the locations).
|
|
2 Discussion
2.1 Marker allele diversity
High levels of polymorphism were observed for the markers, with a range of 2~16 alleles and average of 6.1 alleles per marker locus, which indicated that the diversity of wheat accessions in this study was relatively high. The above results also indicated that EST-SSR markers in this study had lower levels of polymorphism as compared to SSR markers. This suggested that genomic-SSR marker is more powerful than EST-SSR marker in evaluation of genetic polymorphism, Similar results were obtained by Leigh et al. (2003) in 56 old and new varieties of bread wheat on the UK Recommended List.
2.2 Linkage disequilibrium
The presence of LD is a prerequisite for association mapping. Some factors increase LD, such as mutation, mating system, genetic isolation, selection and so on. In the other hand, factors such as recurrent mutations, high recombination and mutation rate decrease LD (Gupta et al. 2005). Besides, the presence of rare alleles can also affect LD to lead inaccurate estimates (Abdurakhmonov and Abdukarimov, 2008). Hence, in this study, markers with rare alleles were replaced with missing values for LD analysis and association mapping. The background LD, which was defined as the distribution of LD between unlinked loci, must be considered to evaluate the extent of LD. Thus, in this study the 95th percentile of the distribution of unlinked r2 estimates was used as baseline LD, instead of r2 = 0.1 or r2 = 0.2 used in previous studies (Palaisa et al. 2003).
Our results showed that LD on chromosome 4A extended up to ~3 cM with r2=0.054 in 103 wheat accessions surveyed with 31 SSR markers. While previous studies indicated that LD was different in different populations of wheat. Maccaferri et al. (2004) reported that LD extended up to 10 and 20 cM with D’= 0.67 in a collection with 134 durum wheat accessions. Somers et al. (2007) reported that LD extended approximately 2~3 cM with r2>0.2, but few loci at longer distances showed high levels of LD (r2 = 0.7 and 1.0) extending 25.5 and 41.2cM. Tommasini et al. (2007) reported that LD on chromosome 3B extended up to 0.5 cM in 44 wheat varieties or 30 cM in 240 RIL populations of winter wheat based on 91 SSR and STS markers. Rhone et al. (2007) found a strong LD extending up to 20 cM with r2 >0.6. Crossa et al. (2007) reported that the r2 declined within ~40 cM below critical values, which occurred at a larger distance than in previous studies in wheat. Probably there are at least three likely causes for the difference of LD extent. The first one is that higher diversity can cause shorter LD extent, because of the many recombination events that have accumulated. In agreement with that, Tenaillon et al. (2001) reported that the extent of LD in nine U.S. inbred lines was higher than in 16 exotic landraces. The second one is that different chromosome has different LD level. For example, Breseghello and Sorrells (2006) reported that LD decayed within a distance of < 1 cM (2D) and < 5 cM (5A) with r2 = 0.065 in 95 soft winter wheat cultivars. The last one is that different marker type has different LD estimates. Remington et al. (2001) found that SSR loci showed proportionally higher estimates of LD than SNPs.
It was also found that LD decay in wheat was far higher than LD decay found in maize. Remington et al. (2001) reported the rapid decay within 1500 bp to values of r2 < 0.1 when assessed with SSRs. Jung et al. (2006) reported the extent of LD within 500 kb in surveying adh1 locus. Andersen et al. (2007) reported that LD persisted over the length of the phenylalanine ammonia lyase (PAL) gene locus (3.7 kb) with the r2 > 0.2 in a survey of 32 European maize inbred lines. The difference between wheat and maize in LD-decay could be due to mating system. Maize, as a highly outcrossing crop species, has much higher recombination than hexaploid wheat, which is an almost completely self-pollinating species, so rapid genome-wide LD decay was determined in maize.
Three LD blocks were observed in the present study on chromosome 4A. One was in the centromeric region and the other two were in the long arm region. The LD block including gwm637, wmc468, wmc707 and wmc161 within 9.5 cM was much longer than the other two, mostly because that marker density in this region was not high enough, and a large LD block might be chopped into smaller pieces by additional markers. The LD block in the bottom of long arm in present study shared a similar position as that reported by Crossa et al. (2007). LD blocks were also found in the centromeric region of chromosome 5A, including 11 loci within 6 cM in 95 cultivars of soft winter wheat (Breseghello and Sorrels, 2006). LD blocks were detected near or in the centromeric region, probably because recombination is less frequent around the centromere (Jones et al. 2002). LD blocks in the short or long arm region were probably caused by artificial selection in the breeding programs, and that would also account for the apparent reduction in allele diversity in that region (Breseghello and Sorrels, 2006).
2.3 Marker-trait associations
In this study, we detected 31 markers covering 85.7 cM on chromosome 4A, with an average marker interval of 2.4 cM (Figure 2). Some results in this study were supported by previous studies. In this study, wmc491 was detected to be associated with PH and GPS both in HD and SY, wmc48 was associated with GPS in HD and FY. Kirigwi et al. (2007) also identified a QTL for grain yield associated with marker wmc48, and a QTL for PH near the marker wmc491 (Figure 2). Meanwhile, the mapping results in this study were also in agreement with some QTLs reported by Araki et al. (1999) and Börner et al. (2002). In previous studies, QTLs for SL and SPS were detected in the marker interval gwm637-gwm160 on chromosome 4A (Li et al. 2002; Jantasuriyarat et al. 2004; Kumar et al. 2007), while wmc707, wmc283 and wmc262 were located in this interval, and they were significantly correlated to SL and SPS in our study (Figure 2). Furthermore, Crossa et al. (2007) found that a DArt marker, wPt8271 which close to barc70 and barc78, was associated with grain yield using association analysis. In our study, barc70 and barc78 were associated with GPS and TKW, while GPS and TKW are two of the most important factors affecting grain yield.
There are some factors affecting association mapping, including population structure, familial relatedness, LD decay, marker density, rare alleles, phenotype definition, environmental risk factors and statistical methods. Considering all of these factors can induce a high resolution of association mapping. In this study, association mapping based on higher density of molecular markers, larger size of populations, a pioneering statistical analysis, short LD extent and repeated experiments under multiple environments significantly increase the resolution of association mapping. Meanwhile, some of the associated markers were in agreement with previous QTL analysis. This study demonstrated that association mapping can be successfully applied in wheat breeding context for detection of marker-traits associations. Furthermore, association mapping can enhance previous QTL information and provide additional QTL information for marker-assisted selection.
3. Materials and methods
3.1 Plant materials and phenotypic analyses
One hundred and three wheat (Triticum aestivum L.) germplasm accessions from China were chosen for this study (data not shown). All varieties were evaluated for the following six agronomical traits at three locations (Haidian (HD), Fuyang (FY) and Shunyi (SY)) in October of 2005, 2006 and 2007, respectively. Plant height (PH, cm) was calculated as the average height of ten plants measured from soil surface to the tip of spike (awns were excluded). Spike length (SL, cm), spikelets per spike (SPS, No./spike) and grains per spike (GPS, No./spike) were measured as the average value/number of the 15 investigated spikes. Thousand-kernel weight (TKW, g) was measured as the average weight of two independent samples of 1,000 grains from each plot. Spikelets density (SD, No./cm) was scored as SPS divided by SL. All statistical analyses were conducted with the software of EXCEL and SPSS (version 12, SPSS, Chicago).
3.2 Genotypic data
DNA was extracted from the embryo of 10 individual seeds using the CTAB method (Saghai-Maroof et al. 1984) with small modification(Using chloroform:octanol (24:1) extraction for only one time). A total of 116 markers including 76 SSR and 40 EST-SSR markers, which amplify the expected fragments, were chosen from thousands of markers for the analysis (data not shown). Based on the consensus map Ta-SSR-2004 (Somers et al. 2004), 31 markers were located on chromosome 4A, the other markers were distributed across the rest wheat chromosomes. All of those markers were selected and synthesized according to the information available in the GrainGenes database (http://wheat.pw.usda.gov/GG2). PCR was carried out in a final reaction volume of 20μL containing 6μL template DNA, 2μL 10×buffer buffer, 0.5μL 2 units/ μL Taq DNA polymerase, 0.4μL 10mmol dNTPs, and 4μL 1.25μM primers. The PCR were performed 35 or 45 cycles at 940C for 45 s, at different annealing temperature (500C to 650C for different primer sets) for 45 s and 720C for 90 s, and a final extension step at 720C for 10 min. The PCR products were separated by electrophoresis in a 6% polyacrylamide gel.
3.3 Marker polymorphism and Population structure
The software of PowerMarker V3.25 (Liu and Muse 2005) was used to calculate allele number, allele frequency, gene diversity, polymorphism information content (PIC) and gene frequency. Rare alleles (with frequency<5%) in the population were treated as missing data for population structure, linkage disequilibrium analysis and association analysis (Breseghello and Sorrells, 2006).
Eight-nine unlinked marker loci, distributed over all the wheat chromosomes, were chosen to assess the population structure of the collection of 103 accessions using the model-based (Bayesian clustering) method implemented in the software of Structure v2.2 (Pritchard et al. 2000a, 2000b; http://pritch.bsd.uchicago.edu/structure.html). The number of subgroups (K) was set from 2 to 12 based on models characterized by admixture and correlated allele frequencies. For each K, five runs were performed separately, 100,000 iterations and a burn-in period of 100,000 were carried out for each run. A value of K was selected when the estimate of InPr(X|K) peaked in the range of 2 to 12 sub-populations.
3.4 Linkage disequilibrium
Linkage disequilibrium between all pairs of loci was evaluated using the software TASSEL v2.0.1 (Bradbury et al., 2007; http://www.maizegenetics.net/bioinformatics/TASSELindex.htm) by setting 1000 permutations. The LD parameter r2 among loci, which is the squared correlation coefficient between two loci and summarizes both mutational and recombination history, were calculated separately for unlinked loci on different chromosomes and for linked loci on the same chromosome (unlinked r2 and syntenic r2, respectively). The loci were considered to be in significant LD if P < 0.001. The LD decay scatterplots of syntenic r2 against genetic distance on chromosomes 4A was drawn using PowerMarker V3.25. LD decay was calculated according to the method described by Breseghello and Sorrels (2006).
3.5 Marker-trait associations
Association between markers and traits was calculated using a mixed linear model (MLM) method in TASSEL v2.0.1 (Yu et al. 2006). The population structure matrix (Q) obtained from the Structure software as described above and the relative kinship matrix (K matrix) derived from the unlinked marker data estimated by TASSEL v2.0.1 were combined to covariate in the association tests to reduce false positive rate. The significant marker-trait associations were declared by P≤0.01 and the magnitude of the QTL effects were evaluated by r2-marker.
Authors' contributions
The author conducted the major part of this study including experimental design, extraction of the DNA, SSR and EST analysis, data analysis and manuscript preparation. Lixin Wang participated in experimental design and preliminary analysis of data. Ji Yao participated in SSR and EST analysis. Yonglian Zheng and Changping Zhao participated in the development of the project, experimental design, and manuscript preparation. All authors read and approved the final manuscript
Acknowledgement
This research was supported by ‘‘863’’ program from the Chinese Ministry of Science and Technology (No. 2006AA100102 and No. 2009AA101102), Beijing Agricultural Breeding Research Platform ІІ(No. D08070500690801), and 948 Ministry of Agriculture project (No. 2009-Z4).
References
Abdurakhmonov I.Y., and Abdukarimov A., 2008, Application of association mapping to understanding the genetic diversity of plant germplasm resources, Int. J. Plant Genomics, doi:10.1155/2008/574927
Agrama H.A., Eizenga G.C., and Yan W., 2007, Association mapping of yield and its components in rice cultivars, Mol. Breed., 19: 341-356 doi:10.1007/s11032-006-9066-6
Andersen J.R., Zein I., Wenzel G., Krützfeldt B., Eder J., Ouzunova M., and Lübberstedt T., 2007, High levels of linkage disequilibrium and associations with forage quality at a Phenylalanine Ammonia-Lyase locus in European maize (Zea mays L.) inbreds, Theor. Appl. Genet., 114: 307-319 doi:10.1007/s00122-006-0434-8
Araki E., Miura H., and Sawada S., 1999, Identification of genetic loci affecting amylose content and agronomic traits on chromosome 4A of wheat, Theor. Appl. Genet., 98: 977-984 doi:10.1007/s001220051158
Beló A., Zheng P.Z., Luck S., Shen B., Meyer D.J., Li B., Tingey S., and Rafalski A., 2008, Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize, Mol. Genet. Genomics, 279: 1-10 doi:10.1007/s00438-007-0289-y
Börner A., Schumann E., Fürste A., Cöster H., Leithold B., Röder M., and Weber W., 2002, Mapping of quantitative trait loci determining agronomic important characters in hexaploid wheat (Triticum aestivum L.), Theor. Appl. Genet., 105: 921-936 doi:10.1007/s00122-002-0994-1
Bradbury P.J., Zhang Z.W., Kroon D.E., Casstevens T.M., Ramdoss Y., and Buckler E.S., 2007, TASSEL: software for association mapping of complex traits in diverse samples, Bioinformatics, 23:2633-2635 doi:10.1093/bioinformatics/btm308
Breseghello F., and Sorrells M.S., 2006, Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars, Genetics, 172:1165-1177 doi:10.1534/genetics.105.044586
Crossa J., Burgueno J., Dreisigacker S., Vargas M., Herrera-Foessel S. A., Lillemo M., Singh R. P., Trethowan R., Warburton M., Franco J., Reynolds M, Crouch J. H., and Ortiz R, 2007, Association analysis of historical bread wheat germplasm using additive genetic covariance of relatives and population structure, Genetics, 177: 1889-1913 doi:10.1534/genetics.107.078659
Flint-Garcia S.A., Thornsberry J.M., and Buckler E.S., 2003, Structure of linkage disequilibrium in plants, Annu. Rev. Plant. Biol., 54: 357-374 doi:10.1146/annurev.arplant.54.031902.134907
Flint-Garcia S.A., Thuillet A.C., Yu J, Pressoir G, Romero S.M., Mitchell S.E., Doebley J, Kresovich S, Goodman M.M., and Buckler E.S., 2005, Maize association population: a high-resolution platform for quantitative trait locus dissection, Plant J., 44: 1054-1064 doi:10.1111/j.1365-313X.2005.02591.x
Gupta P.K., Rustgi S, and Kulwal P.L., 2005, Linkage disequilibrium and association studies in higher plants: present status and future prospects, Plant Mol. Biol., 57: 461-485 doi:10.1007/s11103-005-0257-z
Harjes C.E., Rocheford T.R., Bai L, Brutnell T.P., Kandianis C.B., Sowinski S.G., Stapleton A.E., Vallabhaneni R, Williams M, Wurtzel E.T., Yan J, and Buckler E.S., 2008, Natural genetic variation in lycopene epsilon cyclase tapped for maize biofortifi cation, Science, 319: 330-333 doi:10.1126/science.1150255
Jantasuriyarat C., Vales M.I., Watson C.J.W., and Lizarazu O.R., 2004, Identification and mapping of genetic loci affecting the free-threshing habit and spike compactness in wheat (Triticum aestivum L.), Theor. Appl. Genet., 108: 261-273 doi:10.1007/s00122-003-1432-8
Jones L.E., Rybka K, and Lukaszewski A.J., 2002, The effect of a deficiency and a deletion on recombination in chromosome 1BL in wheat, Theor. Appl. Genet., 104: 1204-1208 doi:10.1007/s00122-002-0876-6
Jun T.H., Van K, Kim M.Y., Lee H.S., and Walker D.R., 2007, Association analysis using SSR markers to find QTL for seed protein content in soybean, Euphytica, 162: 179-191 doi:10.1007/s10681-007-9491-6
Jung M., Ching A., Bhattramakki D., Dolan M., Tingey S., Morgante M., and Rafalski A., 2004, Linkage disequilibrium and sequence diversity in a 500-kbp region around the adh1 locus in elite maize germplasm, Theor. Appl. Genet., 109: 681-689 doi:10.1007/s00122-004-1695-8
Kirigwi F.M., Ginkel M.V., Brown-Gedira G., Gill B.S., Paulsen G.M., and Fritz A.K., 2007, Markers associated with a QTL for grain yield in wheat under drought, Mol. Breed., 20: 401-413 doi:10.1007/s11032-007-9100-3
Kraakman A.T.W., 2006, Linkage disequilibrium mapping of morphological, resistance, and other agronomically relevant traits in modern spring barley cultivars, Mol. Breed., 17: 41-58 doi:10.1007/s11032-005-1119-8
Kumar N, Kulwal P.L., Balyan H.S., and Gupta P.K., 2007, QTL mapping for yield and yield contribution traits in two mapping populations of bread wheat, Mol. Breed., 19: 163-177 doi:10.1007/s11032-006-9056-8
Lander E.S., and Schork N.J., 1994, Genetic dissection of complex traits, Science, 265: 2037-2048 doi:10.1126/science.8091226
Leigh F., Lea V., Law J., Wolters P., Powell W., and Donini P., 2003, Assessment of EST- and genomic microsatellite markers for variety discrimination and genetic diversity studies in wheat, Euphytica, 133: 359-366 doi:10.1023/A:1025778227751
Li W.L., Nelson J.C., Chu C.Y., Shi L.H., Huang S.H., and Liu D.J., 2002, Chromosomal locations and genetic relationships of tiller and spike characters in wheat, Euphytica, 125:357-366 doi:10.1023/A:1016069809977
Liu, and Muse, 2005, PowerMaker: an integrated analysis environment for genetic maker analysis, Bioinformatics, 21: 2128-2129
Palaisa K.A., Morgante M., Williams M., and Rafalski A., 2003, Contrasting effects of selection on sequence diversity and linkage disequilibrium at two phytoene synthase loci, Plant Cell, 15: 1795-1806 doi:10.1105/tpc.012526
Peng J.H., Bai Y., Haley S.D., and Lapitan N.L.V., 2009, Microsatellite-based molecular diversity of bread wheat germplasm and association mapping of wheat resistance to the Russian wheat aphid, Genetica, 135: 95-122 doi:10.1007/s10709-008-9262-x
Pestsova E., Ganal M.W., and Röder M., 2000, Isolation and mapping of microsatellite markers specific for the D genome of bread wheat, Genome, 43: 689-697 doi:10.1139/gen-43-4-689
Pritchard J.K., Stephens M, and Donnelly P, 2000a, Inference of population structure using multilocus genotype data, Genetics, 155: 945-959
Pritchard J.K., Stephens M, Rosenberg N.A., and Donnelly P, 2000b, Association mapping in structured populations, Am. J. Hum. Genet., 67: 170-181 doi:10.1086/302959
Remington D.L., Thornsberry J.M., Matsuola Y, Wilson L.M., Whitt S.R., Doebley J, Kresovich S, Goodman M.M., and Buckler E.S., 2001, Structure of linkage disequilibrium and phenotypic associations in the maize genome, Proc. Natl. Acad. Sci., USA, 98: 11479-11484 doi:10.1073/pnas.201394398
Rhone B., Raquin A.L., and Goldringer I., 2007, Strong linkage disequilibrium near the selected Yr17 resistance gene in a wheat experimental population, Theor. Appl. Genet., 114: 787-802 doi:10.1007/s00122-006-0477-x
Röder M., Korzun V., Wendehake K., Plaschke J., Tixier M.H., Leroy P., and Ganal M.W., 1998, A microsatellite map of wheat, Genetics, 149: 2007-2023
Somers D.J., Isaac P., and Edwards K., 2004, A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.), Theor. Appl. Genet., 109: 1105-1114 doi:10.1007/s00122-004-1740-7
Somers D.J., Banks T., Depauw R., Stephen F., John C., Pozniak C., and McCartney C., 2007, Genome-wide linkage disequilibrium analysis in bread wheat and durum wheat, Genome, 50: 557-567 doi:10.1139/G07-031
Stich B., Melchinger A.E., Frisch M, Maurer H.P., Heckenberger M, and Reif J.C., 2005, Linkage disequilibrium in European elite maize germplasm investigated with SSRs, Theor. Appl. Genet., 111: 723-730 doi:10.1007/s00122-005-2057-x
Tenaillon M.I., Sawkins M.C., Long A.D., Gaut R.L., Doebley J.F., and Gaut B.S., 2001, Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.), Genetics, 162: 1401-1413
Thornsberry J.M., Goodman M.M., Doebley J., Kresovich S., Nielsen D., and Buckler E.S., 2001, Dwarf8 polymorphisms associate with variation in flowering time, Nat. Genet., 28: 286-289 doi:10.1038/90135
Tommasini L., Schnurbusch T., Fossati D., Mascher F., and Keller B., 2007, Association mapping of Stagonospora nodorum blotch resistance in modern European winter wheat varieties, Theor. Appl. Genet., 115: 697-708 doi:10.1007/s00122-007-0601-6
Weigel D, and Nordborg M., 2005, Natural variation in Arabidopsis. How do we find the causal genes? Plant Physiol., 138: 567-568 doi:10.1104/pp.104.900157
Wen W., Mei H.W., Feng F.J., Yu S.B., Huang Z.C., Wu J.H., Chen L., Xu X.Y., and Luo X.J., 2009, Population structure and association mapping on chromosome 7 using a diverse panel of Chinese germplasm of rice (Oryza sativa L.), Theor. Appl. Genet., 119: 459-470 doi:10.1007/s00122-009-1052-z
Yao J., Wang L.X., Liu L.H., Zhao C.P., and Zheng Y.L., 2009, Association mapping of agronomic traits on chromosome 2A of wheat, Genetica, 137: 67-75 doi:10.1007/s10709-009-9351-5
Yu J., and Buckler E,S, 2006, Genetic association mapping and genome organization of maize, Curr Opin Biotechno, 17: 1-6
Yu J., Pressoir G., Briggs W.H., Bi I.V., Yamasaki M, Doebley J.F., McMullen M.D., Gaut B.S., Nielsen D.M., Holland J.B., Kresovich S., and Buckler E.S., 2006, A unified mixed-model method for association mapping that accounts for multiple levels of relatedness, Nat. Genet., 38: 203-208 doi:10.1038/ng1702
Zwart R.S., Muylle H., Bockstaele E., and Roldán-Ruiz I., 2008, Evaluation of genetic diversity of Fusarium head blight resistance in European winter wheat, Theor. Appl. Genet., 117: 813-828 doi:10.1007/s00122-008-0822-3



. PDF(296KB)
. FPDF(win)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Lihua Liu
. Lixin Wang
. Ji Yao
. Yonglian Zheng
. Changping Zhao
Related articles
. Association mapping
. Population structure
. Linkage disequilibrium
. Mixed linear model
Tools
. Email to a friend
. Post a comment