Research Report
A unified framework for causal inference in statistical genetics: integrating GWAS, molecular QTL, colocalization, and Mendelian randomization 
 Author
Author  Correspondence author
Correspondence author
Plant Gene and Trait, 2026, Vol. 17, No. 3 doi: 10.5376/pgt.2026.17.0011
Received: 30 Mar., 2026 Accepted: 26 Apr., 2026 Published: 20 May, 2026
Fang X.J., 2026, A unified framework for causal inference in statistical genetics: integrating GWAS, molecular QTL, colocalization, and Mendelian randomization, Plant Gene and Trait, 17(3): 156-172 (doi: 10.5376/pgt.2026.17.0011)
Genome-wide association studies (GWAS) have identified thousands of loci associated with complex traits, yet translating these statistical signals into biological mechanisms remains a major challenge. A key difficulty lies in distinguishing between association, shared genetic architecture, and causal relationships across multiple layers of molecular regulation. In this study, we present a unified analytical framework for causal inference in statistical genetics that integrates GWAS, molecular quantitative trait loci (QTL), transcriptome-wide association studies (TWAS), colocalization analysis, and Mendelian randomization (MR). Within this framework, different methods address distinct inferential targets: GWAS identifies variant–trait associations; molecular QTL and TWAS link genetic variation to intermediate phenotypes; colocalization evaluates the consistency of signals across datasets; and MR estimates the direction and magnitude of potential effects under explicit assumptions. We emphasize that these components should not be interpreted in isolation but as part of a sequential process of evidence refinement. In particular, colocalization is necessary for prioritizing candidate mechanisms but does not establish causality, while MR provides effect estimates that remain sensitive to instrument validity, pleiotropy, and data heterogeneity. We further discuss practical considerations for implementation, including instrument selection, diagnostic evaluation, and cross-population validation, as well as challenges arising from pleiotropy, tissue specificity, and environmental interactions. Finally, we extend this framework to plant systems and emerging multi-omics contexts, highlighting the role of single-cell and epigenomic data in refining causal interpretation. By clarifying the roles and limitations of individual methods within an integrated framework, this study provides a structured approach for moving from genetic associations toward biologically interpretable and experimentally testable hypotheses.
1 Introduction
Genome-wide association studies (GWAS) have generated an unprecedented scale of statistical associations in complex trait genetics. However, these signals fundamentally represent association estimands-statistical relationships between genetic variants and traits-rather than direct evidence of biological causality. This distinction underlies a central gap in the field: many GWAS loci reside in non-coding regions and are influenced by linkage disequilibrium (LD) and multilayer regulatory architectures, such that a single association peak often implicates multiple candidate genes and mechanisms. Even when fine-mapping reduces signals to smaller credible sets, the resulting inference remains a causal probability estimand, rather than a direct estimate of causal direction or effect (Liu et al., 2019; Wainberg et al., 2019; Xie et al., 2021; Mostafavi et al., 2023).
From a unified statistical genetics perspective, the analysis of complex traits can be conceptualized as a multi-layer inferential chain defined by distinct statistical targets (estimands): GWAS characterizes association evidence, fine-mapping quantifies posterior probabilities of causality, and polygenic risk scores (PRS) translate these signals into individual-level predictive functionals. Yet a critical gap remains in this chain: how to move from causal probability to causal pathways and causal effect estimands. This gap defines the role of functional integration and causal inference methods.
To bridge this divide, expression quantitative trait loci (eQTL) and transcriptome-wide association studies (TWAS) introduce molecular phenotypes as intermediate layers, extending inference from the variant level to the gene expression level. In this framework, eQTL analyses characterize the mapping from genotype to gene expression, while TWAS integrates GWAS summary statistics with expression prediction models to generate gene-level association signals and prioritize candidate genes (Wainberg et al., 2019; Xie et al., 2021; Zhao et al., 2022).
However, it is essential to emphasize that, TWAS remains an association-based projection under LD structure, not a causal estimand. Due to co-regulation among nearby genes, LD contamination, tissue mismatch, and genetic confounding, TWAS signals may reflect non-causal variants, leading to the prioritization of “bystander genes” (Liu et al., 2019; Zhao et al., 2022; Tambets et al., 2024). Thus, TWAS provides necessary but insufficient evidence for causality, and its statistical target remains a gene-level reparameterization of the association estimand.
Colocalization analysis provides a critical interface at this stage. Rather than constituting an independent association test, colocalization evaluates whether GWAS and molecular QTL signals share the same underlying causal variant by comparing their posterior distributions. In this sense, colocalization can be understood as an inference on shared causal configuration estimands derived from fine-mapping posterior distributions. This probabilistic framework enables the integration of association and functional evidence across data domains.
Building upon this structure, Mendelian randomization (MR) advances inference from causal probability to causal effect estimands. By using genetic variants as instrumental variables (IVs), MR estimates the causal effect of an exposure (e.g., gene expression or protein level) on an outcome trait, thereby mitigating confounding and reverse causation under three core assumptions: relevance, independence, and exclusion restriction (Hemani et al., 2018; Jiang et al., 2022).
In practice, however, MR is subject to several challenges, including horizontal pleiotropy, complex LD structure, and weak instruments, all of which can introduce bias and inflate false positives (Barfield et al., 2018; Tambets et al., 2024). Moreover, the limited availability of strong and cross-tissue stable cis-eQTL instruments constrains the applicability and reproducibility of MR in causal gene identification (Lu et al., 2024). Recent methodological developments-such as MR-Egger, weighted median estimators, and MR-PRESSO-provide partial robustness to these violations, but their validity critically depends on the structural evidence provided by upstream eQTL/TWAS and colocalization analyses (Hemani et al., 2018; Zhao et al., 2022).
Based on these considerations, we propose a unified causal inference layer in statistical genetics, in which different methods correspond to distinct estimands within a coherent inferential hierarchy:
-GWAS: association estimand
-Fine-mapping: causal probability estimand
-eQTL/TWAS: mediation mapping estimand
-Colocalization: shared causal configuration estimand
-MR: causal effect estimand
Within this framework, complex trait analysis can be formalized as a “functionally integrated causal chain”: GWAS → fine-mapping → eQTL/TWAS → colocalization → MR. This pipeline represents a progressive refinement from statistical association to causal effect estimation, where each layer is defined by its estimand, assumptions, and sources of uncertainty. This estimand-driven perspective can also be extended to multi-trait genetics, where shared genetic architecture is distinguished from causal interpretation through structural, locus-level, and pattern-level inference (Fang, 2026).
In this study, we systematically develop this unified framework by clarifying the statistical foundations, assumptions, and limitations of each component, with particular emphasis on their interfaces and error propagation across layers. We further propose an operational workflow and reporting standards applicable to multi-ancestry and multi-tissue settings. By reframing functional integration as an estimand-driven inference system rather than a collection of tools, this framework establishes a coherent theoretical foundation for bridging association discovery, causal probability, and mechanistic interpretation in complex trait genetics.
2 Integration of eQTL with Functional Phenotypes
In studies of complex traits, eQTL analyses provide an essential intermediate layer that links genetic variation to molecular phenotypes. Unlike GWAS, which captures statistical associations between loci and traits, eQTL focuses on how genetic variants influence gene expression or related molecular traits, thereby offering clues about potential regulatory mechanisms. In this sense, eQTL analyses do not by themselves establish causality, but instead help delineate the pathways through which genetic variants may act.
Within integrative frameworks, eQTL data are typically used in two ways. First, they provide molecular signals that can be compared with GWAS results through colocalization analyses. Second, they serve as a source of candidate instruments for downstream causal inference methods, such as Mendelian randomization. As such, eQTL analyses form a critical bridge between statistical association and functional interpretation.
2.1 cis-eQTL and trans-eQTL
eQTL are commonly categorized into cis- and trans-acting variants based on their genomic proximity to the target gene. Cis-eQTL are located near the gene they regulate and typically exert their effects through local regulatory elements such as promoters, enhancers, or untranslated regions. Large-scale datasets across multiple tissues have shown that cis-regulatory effects are widespread and relatively reproducible, with a substantial proportion overlapping GWAS loci (Liu et al., 2019; Wainberg et al., 2019). For this reason, cis-eQTL are often prioritized in integrative analyses as plausible links between genetic variants and gene expression.
In practice, cis-eQTL play two major roles. They can be used in colocalization analyses to assess whether GWAS and expression signals are likely driven by the same underlying variant. They also tend to provide more stable instruments for Mendelian randomization, enabling the evaluation of relationships between gene expression and phenotypic traits. At the same time, interpretation requires caution. Linkage disequilibrium may cause signals to spread across neighboring variants, and allelic heterogeneity can complicate the assignment of effects to specific loci. It is therefore common to combine eQTL results with fine-mapping, allele-specific expression, and functional annotations to strengthen the evidence.
Trans-eQTL, in contrast, affect genes located at a distance and often operate through indirect regulatory mechanisms, such as transcription factors, microRNAs, or chromatin interactions (Kirsten et al., 2015). These effects are typically weaker and more sensitive to cellular composition, environmental influences, and population structure, which makes their detection and interpretation more challenging. Recent work suggests that some trans effects arise through hierarchical regulatory relationships, where a cis-regulated gene acts as an upstream driver influencing downstream targets (Kvamme et al., 2025). This observation has practical implications for analysis strategies. One approach is to first identify candidate cis-regulatory variants, then explore their downstream impact using network-based methods or mediation analyses. In some cases, stepwise Mendelian randomization can be applied to evaluate relationships across multiple layers (Figure 1).
|
Figure 1 Regulatory patterns of cis- and trans-eQTL and their roles in integrative analyses |
.png)
In plant systems, these challenges are often amplified by extended linkage disequilibrium, structural variation, and polyploid genomes. Incorporating multi-parent populations (such as NAM or MAGIC) and pangenome references can help reduce misattribution and improve robustness.
Image caption: Cis-eQTLs typically influence gene expression through proximal regulatory elements, such as promoters or enhancers, and are therefore more readily aligned with GWAS signals in integrative analyses. In contrast, trans-eQTLs act on distal genes through indirect mechanisms involving transcription factors, miRNAs, or chromatin interactions, often reflecting multi-layer regulatory processes and increased sensitivity to cellular and population context. The figure illustrates the differences in regulatory scope and pathways between these two classes, highlighting the transition from local regulatory effects to broader network-level influences.
2.2 Tissue and cell-type specificity
eQTL effects often vary substantially across tissues and developmental stages (Fagny et al., 2017). Some loci exhibit consistent effects across multiple tissues, whereas others are highly tissue-specific, reflecting differences in chromatin states, regulatory programs, and cellular composition. For integrative analyses, prioritizing tissues that are relevant to the trait of interest generally improves interpretability. At the same time, reporting both shared and tissue-specific effects can help distinguish robust signals from context-dependent ones.
Cellular heterogeneity represents an important source of confounding in bulk tissue data. To address this, approaches such as deconvolution, interaction models, and environment-specific analyses can be used to better characterize context-dependent effects (Zhang and Zhao, 2023).
Single-cell eQTL (sc-eQTL) analyses further increase resolution by identifying regulatory effects at the level of individual cell types or states. Studies in immune and brain tissues have revealed a large number of cell-type-specific signals, as well as dynamic changes across conditions (Bryois et al., 2022). From a statistical perspective, pseudo-bulk aggregation and hierarchical modeling are often used to balance resolution and power. These data can provide more precise information about where regulatory effects are likely to act, which is valuable for downstream interpretation.
2.3 Extended QTL types: sQTL, pQTL, and meQTL
Beyond expression QTL, additional layers of molecular QTL provide further insight into regulatory mechanisms. Splicing QTL (sQTL) capture variation in transcript structure and may operate independently of total expression levels, allowing regulatory effects to be examined from both quantitative and structural perspectives (Zheng et al., 2020). Analyses at the transcript level or using multi-exposure models can help disentangle these contributions.
Protein QTL (pQTL) measure the effects of genetic variants on protein abundance and are often closer to the functional endpoints of many traits. While some cis-pQTL can serve as strong instruments, their relationship with eQTL is not always straightforward, reflecting additional layers of post-transcriptional regulation.
Methylation QTL (meQTL) describe the influence of genetic variation on DNA methylation patterns. When combined with data on chromatin accessibility and histone modifications, they contribute to a broader view of regulatory architecture. In multi-tissue settings, meQTL signals often only partially overlap with eQTL or GWAS loci, suggesting that different regulatory layers may act through distinct pathways.
In plant systems, particular attention should be given to differences among methylation contexts (CG, CHG, and CHH) and their regulatory mechanisms. Incorporating tissue- or environment-specific analyses can improve the interpretability of these signals.
3 TWAS: From GWAS to Gene-Level Associations
Within integrative analyses, transcriptome-wide association studies (TWAS) provide a framework for translating GWAS signals from the variant level to the gene level. By incorporating gene expression as an intermediate phenotype, TWAS enables the evaluation of whether genetically regulated expression is associated with complex traits, thereby offering a structured link between genetic variation and downstream phenotypes.
It is important to note, however, that TWAS does not directly establish causality. Rather, it reorganizes SNP-level association signals into gene-level statistics under a specified expression prediction model and LD structure. In this sense, TWAS is best viewed as a structured projection of GWAS signals, rather than an independent causal inference approach.
3.1 Basic principles
The central idea of TWAS is to use reference datasets that contain both genotype and expression data to train predictive models of gene expression, and then apply these models to GWAS data to assess gene–trait associations (Li and Ritchie, 2021; Evans et al., 2024). For a given gene g, the predicted expression can be written as:
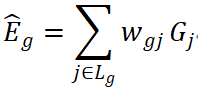
where Lg typically denotes SNPs within a cis region, wgj represents weights estimated from reference data (e.g., using Elastic Net, BLUP, or BSLMM), and Gj denotes SNP dosage. Association is then tested between 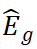 and the phenotype Y, with the goal of evaluating whether genetically driven variation in expression is related to the trait (Mai et al., 2023).
and the phenotype Y, with the goal of evaluating whether genetically driven variation in expression is related to the trait (Mai et al., 2023).
When only GWAS summary statistics are available, TWAS can be implemented using LD-based transformations. Let ![]() denote the vector of SNP-level GWAS Z-scores and
denote the vector of SNP-level GWAS Z-scores and ![]() the corresponding LD matrix. The gene-level statistic can be approximated as:
the corresponding LD matrix. The gene-level statistic can be approximated as:
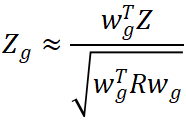
The significance of this statistic depends on the effective information carried by the weights under the LD structure. Two practical considerations follow from this formulation. First, the LD reference panel should closely match the target GWAS population. Second, the choice of tissue or cell type used to train the expression model plays a critical role in determining both interpretability and effect direction (Li and Ritchie, 2021).
3.2 Common methods
Most TWAS methods follow a two-step strategy involving model training and downstream association testing, but differ in how expression weights are constructed and how multi-tissue information is incorporated. PrediXcan and its summary-based extension S-PrediXcan use elastic net regression to estimate cis-regulatory weights in reference data, which are then applied to individual-level or summary-level GWAS data. MultiXcan and UTMOST extend this framework by integrating information across tissues to distinguish shared from tissue-specific effects.
FUSION adopts a more flexible approach by integrating multiple prediction models-including BLUP, LASSO, Elastic Net, and BSLMM-within a unified framework, allowing direct computation of gene-level statistics from summary data while accounting for model uncertainty and LD structure (Evans et al., 2024). This class of methods emphasizes robustness in model selection and statistical inference.
More recent developments include approaches that incorporate nonparametric or Bayesian modeling strategies (e.g., TIGAR and its extensions), as well as methods that integrate multiple priors to improve power (Parrish et al., 2022; Liang et al., 2025). These extensions broaden the applicability of TWAS across diverse data settings.
In practice, different methods reflect distinct priorities. Some emphasize the portability of expression weights across datasets, whereas others focus on model integration and uncertainty control. Regardless of the approach, TWAS results are typically interpreted in conjunction with fine-mapping and colocalization analyses, which help refine gene-level signals into more credible candidate regions (Li and Ritchie, 2021; Mai et al., 2023).
3.3 Limitations
Despite its utility in linking GWAS signals to functional interpretation, TWAS has several important limitations. First, the transferability of expression prediction models is often constrained. The estimated weights depend on the ancestry, LD structure, and tissue context of the reference dataset. When these differ from those of the target GWAS population, prediction accuracy may decline, leading to reduced statistical power and potential bias (Li and Ritchie, 2021; Mai et al., 2023). Multi-tissue approaches and expanded reference resources such as GTEx can mitigate this issue to some extent, but do not fully resolve it.
Second, TWAS results remain fundamentally associative. Because of LD, the weights used to predict expression may capture signals from variants that are correlated with, but not identical to, the true causal variant. In addition, co-regulation and unobserved confounding can cause non-causal genes to appear significantly associated. As a result, interpreting TWAS findings as evidence of causal effects can be misleading (Wainberg et al., 2019; Evans et al., 2024). Simulation and methodological studies have shown that such interpretations may inflate false positive rates if not carefully controlled (Zhu and Zhou, 2020; De Leeuw et al., 2023).
For this reason, TWAS findings are typically evaluated alongside locus-level evidence. Colocalization analyses can be used to assess whether GWAS and expression signals are consistent with a shared underlying variant, while Mendelian randomization can provide additional support for potential causal relationships. This layered approach helps reduce misinterpretation and improves the reliability of downstream inference.
Finally, the scope of TWAS is limited by the availability and coverage of reference datasets. Current eQTL resources provide incomplete representation of rare variants, trans-regulatory effects, and noncoding RNAs, which constrains the comprehensiveness of the models. Future developments are likely to focus on more flexible modeling strategies, expanded multi-ancestry and multi-tissue datasets, and explicit modeling of genotype-by-environment interactions, particularly in plant and multi-environment studies (Parrish et al., 2022; Liang et al., 2025).
4 The Role and Limitations of Colocalization Analysis
In moving from GWAS signals toward functional interpretation, a central question is whether association signals observed in different data sources-such as GWAS and molecular QTL-reflect the same underlying genetic factors within a given genomic region. Colocalization analysis was developed to address this question by evaluating the consistency of signals across datasets and providing a basis for downstream interpretation.
Unlike association analysis within a single dataset, colocalization focuses on the correspondence between signals from different sources. The goal is to assess whether two signals can plausibly be explained by the same underlying variant, given the local LD structure and statistical uncertainty. In practice, this step serves to refine candidate regions, narrowing the focus from general associations to loci that are more likely to support coherent biological interpretation.
4.1 Statistical framework of colocalization
Widely used approaches such as COLOC adopt a Bayesian framework to compare a set of mutually exclusive hypotheses, including no association, association in only one dataset, independent associations in both datasets, and a shared underlying variant. Posterior probabilities are assigned to each scenario, with PPH4 commonly used as a summary measure of support for the shared-variant hypothesis (Zuber et al., 2022).
As genomic regions often contain multiple independent signals, the assumption of a single causal variant is frequently violated. To address this, more recent methods incorporate multi-signal modeling and integrate colocalization with fine-mapping, such as coloc combined with SuSiE or FINEMAP, as well as approaches like eCAVIAR and fastENLOC (Foley et al., 2021; Wallace, 2021). These developments improve performance in regions with complex signal structures and allow colocalization to be more closely aligned with locus-level inference.
In practice, reliable colocalization requires careful data harmonization. This includes aligning allele orientation, standardizing effect sizes and standard errors, and using LD reference panels that match the ancestry of the GWAS dataset. Because multiple independent signals may be present within a region, it is often advisable to perform conditional analysis or fine-mapping prior to colocalization, or to apply models that explicitly account for multiple signals. In multi-tissue settings, analyses can be conducted separately for each tissue and then integrated with functional annotations to form a more complete interpretation (Wallace, 2021; Zuber et al., 2022).
Although thresholds such as PPH4 > 0.8 are often used in practice, their interpretation depends on model assumptions, prior choices, and data quality. Sensitivity analyses and conditional results should therefore be considered when evaluating the robustness of findings (Figure 2) (Rasooly et al., 2022).
|
Figure 2 Statistical decision framework of colocalization analysis and its role in cross-dataset integration Image caption: Colocalization analysis evaluates whether association signals from GWAS and molecular QTL (e.g., eQTL) within the same genomic region are consistent with a shared underlying genetic variant. Under a Bayesian framework, methods such as COLOC compare five mutually exclusive scenarios, including no signal, GWAS-only, QTL-only, independent signals, and shared signals, thereby quantifying alternative explanations for the observed patterns. The figure illustrates these scenarios and the corresponding signal configurations, with PPH4 representing the posterior support for the shared-variant hypothesis. It should be noted that PPH4 reflects statistical evidence for signal concordance rather than direct inference of causal mechanisms or mediation |
.png)
4.2 Interpreting colocalization results
While colocalization is a useful tool for prioritizing candidate loci, its results should not be interpreted as direct evidence of causal relationships. Even when the posterior probability for a shared signal is high, this only indicates that two associations are consistent with the same underlying variant; it does not establish how that variant influences the trait.
One common scenario is horizontal pleiotropy, in which a single variant affects multiple traits through independent pathways. In such cases, colocalization may still detect a shared signal, even though no direct mediation relationship exists between the molecular phenotype and the complex trait (Rasooly et al., 2022). Evidence from epigenomic studies further suggests that many disease-associated loci involve complex regulatory architectures with multiple parallel pathways (Shikov et al., 2020; Boix et al., 2021; Khan et al., 2024).
LD structure and allelic heterogeneity can also complicate interpretation, particularly in regions with multiple signals where the true causal variant may be obscured by correlated variants (Wallace, 2021). For these reasons, colocalization is best viewed as a filtering step rather than a definitive test.
In analytical workflows, loci with strong colocalization support are often prioritized for further evaluation using complementary approaches. For example, Mendelian randomization can be applied to assess the direction and magnitude of potential effects, whereas loci with weak or inconsistent evidence may require re-examination at the level of fine-mapping or data harmonization before further interpretation.
4.3 Applications in plant systems
In plant systems, colocalization analysis has proven useful for disentangling tissue-specific and environment-dependent regulatory effects. Studies across multiple tissues and environmental conditions have shown that the effects of regulatory variants can vary substantially depending on developmental stage or external stimuli, leading to context-dependent contributions to complex traits.
For instance, analyses in crops and model plants have identified distinct regulatory patterns across organs such as leaves, roots, and fruits, as well as across developmental stages. Work in tomato and other species has further demonstrated that conserved developmental genes may exhibit pleiotropic effects, while still showing variation in regulatory behavior across environments or species (Hendelman et al., 2021).
In practice, it is often beneficial to construct eQTL maps in trait-relevant tissues and under representative environmental conditions, followed by stratified colocalization analyses across these contexts. The use of multi-parent populations, such as NAM or MAGIC, can improve resolution by reducing LD and helping to distinguish multiple signals. In polyploid crops, additional attention is required to differentiate homologous gene copies and to accurately quantify expression, in order to minimize ambiguity in gene assignment.
Candidate loci identified through colocalization can then be further evaluated using downstream approaches, including Mendelian randomization, near-isogenic lines, genome editing, and expression assays, ultimately contributing to a more complete characterization of the relationship between genetic variation and phenotypic traits.
5 Mendelian Randomization as A Framework for Causal Inference
Within integrative analysis pipelines, Mendelian randomization (MR) is typically applied at a later stage, once genetic associations and molecular evidence have been established. Its primary purpose is to evaluate the direction and magnitude of potential effects by using genetic variants as instruments and treating molecular traits-such as gene expression-as exposures.
Compared with earlier steps, which focus on mapping or aligning signals across datasets, MR aims to quantify relationships under a set of assumptions. As a result, the interpretation of MR findings depends critically on how instruments are selected and on the plausibility of the underlying assumptions.
5.1 Core assumptions of instrumental variables
MR analyses rely on three basic conditions. First, the selected genetic variants must be sufficiently associated with the exposure, ensuring that they carry informative signal. Second, these variants should be independent of confounding factors, an assumption that is partly justified by the approximate random allocation of alleles at conception. Third, the effect of the instruments on the outcome should operate primarily through the exposure of interest, rather than through alternative pathways.
In practice, these conditions are not directly testable and may be violated in subtle ways. For example, a variant may influence multiple biological processes, giving rise to additional pathways that complicate interpretation. For this reason, instrument selection is often informed by upstream analyses. Colocalization, in particular, can be used to identify regions where GWAS and molecular QTL signals are consistent with a shared underlying variant, thereby increasing confidence that the selected instruments reflect a common source of variation (Zuber et al., 2022).
5.2 Common estimation approaches
Among available methods, inverse-variance weighted (IVW) regression is most commonly used as the primary estimator. It combines ratio estimates from multiple instruments-defined as the effect of a variant on the outcome divided by its effect on the exposure-using inverse-variance weighting. When all instruments are valid, or when biases average out, IVW provides efficient estimates. In the case of a single instrument, the Wald ratio is typically used.
When heterogeneity is present across instruments, random-effects formulations can be applied to account for additional variance. A number of complementary approaches have been developed to improve robustness. MR-Egger regression introduces an intercept term to detect directional bias and, under certain conditions, provides a corrected estimate, although at the cost of reduced precision. The weighted median estimator remains consistent when a subset of instruments is invalid, while mode-based estimators offer further robustness under specific assumptions about the distribution of effects.
In settings where multiple exposures may contribute jointly-such as gene expression, splicing, or protein abundance-multivariable MR (MVMR) can be used to disentangle their contributions. Although this approach can provide more detailed insight into complex regulatory relationships, it also requires stronger assumptions and higher data quality.
5.3 Weak instruments and pleiotropy
The strength of the instruments plays a central role in determining the stability of MR estimates. Weak associations between instruments and exposure can lead to biased estimates that are closer to observational correlations, along with inflated uncertainty. The F statistic is commonly used as a diagnostic measure, with a value of around 10 often considered a practical benchmark in univariable analyses. In multivariable settings, instrument strength needs to be evaluated separately for each exposure.
When weak instruments are detected, several strategies may help improve performance, including applying stricter selection thresholds, restricting analyses to cis-regulatory variants, or using reference datasets that better match the target population. Issues such as sample overlap and winner’s curse can further weaken instruments and should be considered during study design and interpretation.
Pleiotropy introduces an additional layer of complexity. Overall heterogeneity can be assessed using statistics such as Cochran’s Q, while the MR-Egger intercept provides a test for directional bias. MR-PRESSO offers procedures for identifying and correcting outlier instruments, and radial MR provides a useful visualization framework for detecting influential points. Sensitivity analyses, including leave-one-out procedures and directionality checks such as the Steiger test, can further help evaluate the robustness of the results.
In reporting, it is generally advisable to consider multiple diagnostics together. When estimates from different methods (e.g., IVW, weighted median, MR-Egger) are broadly consistent and diagnostic tests do not indicate major violations, the findings are more likely to be reliable. When discrepancies arise, it is often necessary to revisit earlier steps, including instrument selection, data harmonization, and upstream evidence.
5.4 Position within integrative analyses
In a broader analytical workflow, MR is typically applied after an initial round of signal prioritization. Colocalization can be used to identify loci where signals from different data sources are consistent, thereby guiding the selection of instruments. MR is then used to further evaluate whether these signals are compatible with a directional relationship between a molecular trait and a complex phenotype.
For loci with stronger supporting evidence, replication across independent datasets or conditions can help assess robustness. In multi-tissue or multi-omics settings, multivariable models may provide additional insight into overlapping pathways. Conversely, when evidence is limited or inconsistent, it is often more appropriate to return to earlier stages of analysis, such as fine-mapping or data harmonization, rather than proceeding directly to causal interpretation.
Overall, MR is best understood as part of a sequence of analytical steps rather than a standalone method. When combined with association analyses, colocalization, and functional annotation, it contributes to a gradual refinement of evidence, moving from statistical association toward more directed interpretation of genetic effects (Zuber et al., 2022).
6 An Integrated Pathway for Causal Inference
In studies of complex traits, different data types and analytical approaches each provide only partial information. Effective integration therefore requires a coherent analytical path through which initial genetic associations can be progressively refined into more interpretable results. Rather than functioning as independent tools, these methods operate in sequence, with each step narrowing the set of candidates and adding complementary evidence.
In practice, analyses often begin with GWAS signals and proceed by incorporating molecular and statistical constraints, gradually focusing from broad genomic regions to more specific genes or regulatory mechanisms.
6.1 From GWAS signals to candidate genes
Analyses typically start with GWAS summary statistics. Initial steps include harmonizing allele orientation, standardizing effect sizes, and selecting LD reference panels matched to the study population. Fine-mapping can then be applied to reduce the set of candidate variants and concentrate the analysis on more localized regions (Hormozdiari et al., 2016).
At this stage, incorporating molecular QTL data provides an additional layer of information. Colocalization analysis is used to assess whether GWAS and molecular signals within a region are consistent with a shared underlying variant, thereby helping to prioritize loci for further investigation. When such consistency is observed, expression-based models can be used to translate locus-level signals into gene-level associations, further narrowing the list of candidate genes (Porcu et al., 2019; Wainberg et al., 2019; Zhang et al., 2024).
Following this prioritization, selected variants can be used as instruments to evaluate relationships between molecular traits and complex phenotypes. Mendelian randomization is commonly applied at this stage to examine potential directionality and estimate effect sizes. Through this sequence, initial GWAS signals are progressively refined into more specific hypotheses, such as the involvement of particular genes or regulatory processes (Lessard et al., 2024).
In situations where data are incomplete-for example, when high-quality eQTL data are unavailable-the analytical path may need to be adapted. TWAS can provide an initial prioritization of candidate genes, which can later be revisited using external or newly generated datasets. In such cases, however, interpretation should remain cautious, particularly when moving toward causal claims (Wainberg et al., 2019).
Across the entire process, replication in independent populations or environments can help assess robustness. Combining multiple MR methods and diagnostic measures further contributes to a more comprehensive evaluation of the evidence (Porcu et al., 2019; Zuber et al., 2022).
6.2 Practical considerations in analysis
In applied settings, the choice of analytical strategy depends on data availability and quality. A key consideration is whether high-quality molecular QTL data are available in tissues relevant to the trait of interest, along with appropriate LD reference panels. When these conditions are not met, the scope of interpretation becomes more limited.
During candidate prioritization, locally acting signals are often easier to interpret and are therefore commonly prioritized. In contrast, distal regulatory signals typically require additional supporting evidence, such as network-based analyses or multi-step approaches, to support interpretation.
Functional annotation also plays an important role. When candidate variants align with known regulatory features-such as open chromatin regions or transcription factor binding sites-the biological plausibility of the findings is strengthened. Conversely, when evidence is limited or multiple signals coexist within a region, additional refinement or data integration may be required (Hormozdiari et al., 2016).
Under favorable conditions-where molecular QTL signals align with GWAS results and suitable instruments can be identified-further analyses can be carried out, with primary estimates reported alongside sensitivity analyses using complementary methods (Porcu et al., 2019; Zuber et al., 2022). When evidence is inconsistent, however, it is often preferable to revisit earlier steps rather than proceed with interpretation.
The final output of such analyses typically combines statistical results with diagnostic measures and functional annotations, allowing candidate genes to be ranked according to the strength of evidence and prioritized for experimental validation (Figure 3) (Votava and Parks, 2021; Lessard et al., 2024).
|
Figure 3 An integrative framework for causal inference in statistical genetics |
.png)
6.3 Examples of application
This integrative approach has been applied across a range of biological systems. In human studies, such as those focusing on lipid-related traits, significant GWAS loci can be examined in relevant tissues (e.g., liver) to identify candidate regions. Colocalization analyses can then be used to prioritize loci showing consistent signals, followed by expression-based analyses and MR to evaluate potential relationships. When results are stable and supported by multiple lines of evidence, they can be further interpreted in the context of known biological pathways or potential therapeutic targets (Porcu et al., 2019; Wainberg et al., 2019; Votava and Parks, 2021; Lessard et al., 2024; Zhang et al., 2024).
In plant systems, similar strategies can be applied to complex traits such as disease resistance. Analyses across tissues and environmental conditions can help identify context-dependent regulatory signals. These can then be integrated with genetic and functional data to prioritize candidate genes. Given the complexity of plant genomes, including extended LD and structural variation, the use of multi-parent populations and pangenome references can improve resolution and interpretation (Zhang et al., 2024).
Together, these examples illustrate that integration across data types is not a matter of simply combining results, but of progressively refining evidence. Through this process, genetic associations can be translated into more specific and testable hypotheses about the mechanisms underlying complex traits.
7 Discussion
7.1 Linking molecular associations with causal evaluation
A central challenge in the study of complex traits lies in connecting genetic associations to mechanistic interpretation. Molecular QTL analyses and TWAS contribute by constraining association signals within a functional context, enabling signals dispersed across the genome to be interpreted at the level of genes or regulatory processes. Mendelian randomization, in turn, builds on this information to evaluate potential directionality and effect magnitude. Rather than acting as independent components, these approaches are linked through intermediate steps-most notably colocalization-which help align signals across datasets and establish continuity between different layers of evidence.
In practice, locally acting regulatory signals, particularly cis-eQTLs derived from trait-relevant tissues, tend to be more stable and interpretable. TWAS further aggregates these signals into gene-level associations, narrowing the pool of candidates. However, such associations remain statistical in nature. Colocalization provides a way to assess whether signals from different sources are compatible with a shared genetic origin, thereby reducing the likelihood of advancing non-causal genes into downstream analyses. Under these conditions, effect estimates obtained from MR become more interpretable in a biological context (Porcu et al., 2019).
With the increasing availability of diverse datasets, this integrative process has extended to multi-tissue and single-cell contexts, allowing regulatory effects to be examined at finer spatial and contextual resolution. For example, cell type–specific regulatory mechanisms have been shown to play a central role in certain disease processes, highlighting the value of incorporating such information into integrative analyses (Gleason et al., 2021).
At the implementation level, the choice of instruments remains a critical factor. Prioritizing cis-regulatory variants, combined with ancestry-matched LD reference panels and tissue-specific models, can improve stability. The use of multiple estimation methods alongside systematic diagnostic procedures further strengthens the robustness of findings (Hemani et al., 2018; Hu et al., 2022). In settings involving multiple molecular layers or tissues, multivariable models can help disentangle overlapping signals and avoid attributing shared regulation to a single pathway (Zuber et al., 2022).
7.2 The impact of pleiotropy and heterogeneity
Despite these advances, integrative analyses remain sensitive to pleiotropy and heterogeneity. Horizontal pleiotropy presents a major challenge, as a single genetic variant may influence multiple traits through independent pathways, complicating interpretation based on a single assumed mechanism. In such cases, effect estimates may deviate from the underlying biological process (Hemani et al., 2018).
Differences across datasets-such as mismatches in tissue relevance, population structure, or environmental context-can further contribute to variability in results. Even when colocalization indicates concordance between signals, this may reflect shared genetic architecture rather than a specific mechanistic pathway (Zuber et al., 2022). Consequently, colocalization should be interpreted as a filtering step rather than as evidence of causality.
Addressing these challenges requires both careful instrument selection and the use of complementary analytical strategies. Restricting analyses to locally acting variants and accounting for LD structure can reduce confounding. At the same time, comparing results across multiple methods helps identify inconsistencies that may indicate violations of assumptions. Diagnostic and sensitivity analyses play a key role in detecting influential or invalid instruments and in assessing the robustness of conclusions (Hu et al., 2022).
Recent methodological developments have sought to model pleiotropy and heterogeneity explicitly, particularly in multi-tissue and multi-context settings. While these approaches show promise, their performance remains dependent on data quality and appropriate model specification (Gleason et al., 2021; Lu et al., 2024).
7.3 Extensions to plant systems
In plant systems, integrative analyses face additional layers of complexity. Environmental effects often play a dominant role, such that the same genetic variant may exhibit different effects across developmental stages, tissues, or stress conditions. In addition, genomic features such as extended LD, structural variation, and polyploidy complicate the interpretation of association signals.
These characteristics necessitate adjustments to analytical strategies. For example, regulatory maps should ideally be constructed under trait-relevant tissues and environmental conditions, with stratified analyses used to capture context-dependent effects. Instrument selection must also account for gene copy number and homology, to avoid ambiguity in signal assignment (Porcu et al., 2019).
Population design is another important consideration. Multi-parent populations can improve resolution by reducing LD and enabling better separation of multiple signals. Replication across environmental conditions helps identify stable regulatory relationships, while multivariable approaches can be used to separate baseline and inducible effects (Lu et al., 2024). Importantly, results that do not support a given hypothesis should also be retained, as they contribute to refining candidate prioritization and avoiding overinterpretation.
7.4 Integration with emerging multi-omics and dynamic systems
The expansion of multi-omics data has made it possible to characterize genetic effects across multiple biological layers. Epigenomic and three-dimensional genome data provide direct evidence for regulatory mechanisms, allowing the physical relationships between variants, regulatory elements, and target genes to be examined. For example, different classes of QTL and chromatin interaction data can jointly describe the connections between enhancers and promoters (Hu et al., 2018; Bhattacharya et al., 2021).
Incorporating these data into integrative frameworks allows for more detailed interpretation of regulatory pathways. When multiple molecular layers-such as expression, splicing, and protein abundance-are considered simultaneously, multivariable approaches can help distinguish their relative contributions.
Single-cell and multimodal data further extend this framework by enabling analyses at the level of cell types and cellular states. These approaches have already revealed highly specific regulatory patterns in several systems, offering new perspectives on the mechanisms underlying complex traits.
Future directions are likely to include the incorporation of temporal and perturbation data, allowing dynamic processes to be modeled more explicitly, as well as network-based approaches that consider groups of genes or regulatory modules. Coupling statistical analyses with high-throughput experimental validation may ultimately lead to integrated systems in which data-driven inference and experimental testing inform one another, advancing the transition from statistical associations to mechanistic understanding and, eventually, to targeted intervention (Colomé-Tatché and Theis, 2018; Bhattacharya et al., 2021).
8 Conclusion
A central challenge in complex trait genetics is not the application of individual methods, but the establishment of a coherent analytical path that connects statistical associations to biological interpretation. In this context, colocalization analysis occupies a critical intermediate position, serving to evaluate the consistency of signals across data sources and to guide the transition from molecular association to downstream inference.
When GWAS and molecular QTL signals show stable correspondence within a genomic region, this provides a basis for prioritizing candidate genes and regulatory elements. However, such evidence reflects compatibility at the level of shared signal rather than direct insight into underlying mechanisms. In other words, colocalization supports entry into further analysis but does not, on its own, establish how a genetic variant influences a trait. Interpreting it as evidence of causality without additional support risks conflating shared genetic architecture with specific biological pathways.
Building on this foundation, Mendelian randomization offers a means to evaluate potential relationships in terms of direction and magnitude. By leveraging genetic variants as instruments, MR extends the analysis from signal alignment toward effect estimation. At the same time, its conclusions remain contingent on a set of assumptions, including the strength and validity of the instruments and the absence of alternative pathways. As a result, MR findings should be interpreted alongside diagnostic measures and complementary methods, and discrepancies should prompt re-examination of earlier analytical steps rather than isolated interpretation.
Taken together, the framework outlined here is best understood not as a fixed pipeline, but as a process of progressive refinement. Starting from GWAS signals, fine-mapping reduces the candidate variant space; molecular QTL data and colocalization analyses help identify signals that are consistent across datasets; expression-based models further narrow the focus to gene-level candidates; and, where appropriate, MR is used to evaluate potential relationships. The outputs of this process extend beyond lists of candidate genes, incorporating levels of supporting evidence and consistency that can guide experimental prioritization.
Although this general strategy is applicable across biological systems, its implementation must be adapted to the characteristics of the data. In human studies, ancestry matching and LD structure play a central role in interpretation, whereas in plant systems, environmental variation, genomic complexity, and gene copy number introduce additional challenges. These differences do not alter the overall framework but influence how individual steps are carried out and weighted.
Looking forward, advances in epigenomics, single-cell technologies, and multimodal datasets will enable the relationships between genetic variation and phenotypic outcomes to be examined across multiple biological layers. Integrating these data into existing analytical frameworks will allow regulatory pathways to be characterized with greater precision. Coupled with high-throughput experimental approaches, such developments have the potential to establish a more continuous link between data-driven inference and mechanistic validation, ultimately advancing the translation of statistical findings into actionable biological insights.
Author Contributions
Xuanjun Fang conducted this study, including literature review, data analysis, and the writing and revision of the manuscript. The author has read and approved the final version of the manuscript.
Acknowledgements
This work was supported by a Major Project of the National Natural Science Foundation of China (Grant No. 30490254).
Barfield R., Feng H., Gusev A., Wu L., Zheng W., Pasaniuc B., and Kraft P., 2018, Transcriptome‐wide association studies accounting for colocalization using Egger regression, Genetic Epidemiology, 42(5): 418-433.
https://doi.org/10.1002/gepi.22131
Bhattacharya A., Li Y., and Love M.I., 2021, MOSTWAS: multi-omic strategies for transcriptome-wide association studies, PLoS Genetics, 17(3): e1009398.
https://doi.org/10.1371/journal.pgen.1009398
Boix C.A., James B.T., Park Y. P., Meuleman W., and Kellis M., 2021, Regulatory genomic circuitry of human disease loci by integrative epigenomics, Nature, 590(7845): 300-307.
https://doi.org/10.1038/s41586-020-03145-z
Bryois J., Calini D., Macnair W., Foo L., Urich E., Ortmann W., Iglesias V.A., Selvaraj S., Nutma E., Marzin M., Amor S., Williams A., Castelo-Branco G., Menon V., De Jager P., and Malhotra D., 2022, Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders, Nature Neuroscience, 25(8): 1104-1112.
https://doi.org/10.1038/s41593-022-01128-z
Colomé-Tatché M., and Theis F.J., 2018, Statistical single cell multi-omics integration, Current Opinion in Systems Biology, 7: 54-59.
https://doi.org/10.1016/j.coisb.2018.01.003
De Leeuw C., Werme J., Savage J.E., Peyrot W.J., and Posthuma D., 2023, On the interpretation of transcriptome-wide association studies, PLoS Genetics, 19(9): e1010517.
https://doi.org/10.1371/journal.pgen.1010921
Evans P., Nagai T., Konkashbaev A., Zhou D., Knapik E.W., and Gamazon E.R., 2024, Transcriptome‐wide association studies (TWAS): methodologies, applications, and challenges, Current Protocols, 4(2): e981.
https://doi.org/10.1002/cpz1.981
Fagny M., Paulson J.N., Kuijjer M.L., Sonawane A.R., Chen C.Y., Lopes-Ramos C.M., Glass K., Quackenbush J., and Platig J., 2017, Exploring regulation in tissues with eQTL networks, Proceedings of the National Academy of Sciences, 114(37): E7841-E7850.
https://doi.org/10.1073/pnas.1707375114
Fang X.J., 2026, A hierarchical inference framework for multi-trait genetics integrating genomic SEM, PLEIO, and Primo, Tree Genetics and Molecular Breeding, 16(1): xx-xx.
Foley C.N., Staley J.R., Breen P.G., Sun B.B., Kirk P.D., Burgess S., and Howson J.M., 2021, A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits, Nature Communications, 12(1): 764.
https://doi.org/10.1038/s41467-020-20885-8
Gleason K.J., Yang F., and Chen L.S., 2021, A robust two‐sample transcriptome‐wide Mendelian randomization method integrating GWAS with multi‐tissue eQTL summary statistics, Genetic Epidemiology, 45(4): 353-371.
https://doi.org/10.1002/gepi.22380
Hemani G., Bowden J., and Davey Smith G., 2018, Evaluating the potential role of pleiotropy in Mendelian randomization studies, Human Molecular Genetics, 27(R2): R195-R208.
https://doi.org/10.1093/hmg/ddy163
Hendelman A., Zebell S., Rodriguez-Leal D., Dukler N., Robitaille G., Wu X., Kostyun J., Tal L., Wang P., Bartlett M.E., Eshed Y., Efroni I., and Lippman Z.B., 2021, Conserved pleiotropy of an ancient plant homeobox gene uncovered by cis-regulatory dissection, Cell, 184(7): 1724-1739.
https://doi.org/10.1016/j.cell.2021.02.001
Hormozdiari F., Van De Bunt M., Segre A.V., Li X., Joo J.W.J., Bilow M., Sul J.H., Sankararaman S., Pasaniuc B., and Eskin E., 2016, Colocalization of GWAS and eQTL signals detects target genes, The American Journal of Human Genetics, 99(6): 1245-1260.
https://doi.org/10.1016/j.ajhg.2016.10.003
Hu X., Zhao J., Lin Z., Wang Y., Peng H., Zhao H., Wang X., and Yang C., 2022, Mendelian randomization for causal inference accounting for pleiotropy and sample structure using genome-wide summary statistics, Proceedings of the National Academy of Sciences, 119(28): e2106858119.
https://doi.org/10.1073/pnas.2106858119
Hu Y., An Q., Sheu K., Trejo B., Fan S., and Guo Y., 2018, Single cell multi-omics technology: methodology and application, Frontiers in Cell and Developmental Biology, 6: 28.
https://doi.org/10.3389/fcell.2018.00028
Jiang L., Miao L., Yi G., Li X., Xue C., Li M. J., Huang H., and Li M., 2022, Powerful and robust inference of complex phenotypes' causal genes with dependent expression quantitative loci by a median-based Mendelian randomization, The American Journal of Human Genetics, 109(5): 838-856.
https://doi.org/10.1016/j.ajhg.2022.04.004
Khan M., Ludl A.A., Bankier S., Björkegren J.L., and Michoel T., 2024, Prediction of causal genes at GWAS loci with pleiotropic gene regulatory effects using sets of correlated instrumental variables, PLoS Genetics, 20(11): e1011473.
https://doi.org/10.1371/journal.pgen.1011473
Kirsten H., Al-Hasani H., Holdt L., Gross A., Beutner F., Krohn K., Horn K., Ahnert P., Burkhardt R., Reiche K., Hackermüller J., Löffler M., Teupser D., Thiery J., and Scholz M., 2015, Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding loci, Human Molecular Genetics, 24(16): 4746-4763.
https://doi.org/10.1093/hmg/ddv194
Kvamme J., Badsha M.B., Martin E.A., Wu J., Wang X., and Fu A.Q., 2025, Causal network inference of cis- and trans-gene regulation of expression quantitative trait loci across human tissues, Genetics, 230(2): iyaf064.
https://doi.org/10.1093/genetics/iyaf064
Lessard S., Chao M., Reis K., FinnGen and Estonian Biobank Research Team, Beauvais M., Rajpal D.K., Sloane J., Palta P., Klinger K., de Rinaldis E., Shameer K., and Chatelain C., 2024, Leveraging large-scale multi-omics evidences to identify therapeutic targets from genome-wide association studies, BMC Genomics, 25(1): 1111.
https://doi.org/10.1186/s12864-024-10971-2
Li B., and Ritchie M.D., 2021, From GWAS to gene: transcriptome-wide association studies and other methods to functionally understand GWAS discoveries, Frontiers in Genetics, 12: 713230.
https://doi.org/10.3389/fgene.2021.713230
Liang Y., Wang H., and Zhang Y.D., 2025, A-TWAS: an aggregated transcriptome-wide association study model incorporating multiple Bayesian priors, bioRxiv, 2025-01.
https://doi.org/10.1101/2025.01.27.635054
Liu B., Gloudemans M.J., Rao A.S., Ingelsson E., and Montgomery S.B., 2019, Abundant associations with gene expression complicate GWAS follow-up, Nature Genetics, 51(5): 768-769.
https://doi.org/10.1038/s41588-019-0404-0
Lu Y., Xu K., Maydanchik N., Kang B., Pierce B.L., Yang F., and Chen L.S., 2024, An integrative multi-context Mendelian randomization method for identifying risk genes across human tissues, The American Journal of Human Genetics, 111(8): 1736-1749.
https://doi.org/10.1016/j.ajhg.2024.06.012
Mai J., Lu M., Gao Q., Zeng J., and Xiao J., 2023, Transcriptome-wide association studies: recent advances in methods, applications and available databases, Communications Biology, 6(1): 899.
https://doi.org/10.1038/s42003-023-05279-y
Mostafavi H., Spence J.P., Naqvi S., and Pritchard J.K., 2023, Systematic differences in discovery of genetic effects on gene expression and complex traits, Nature Genetics, 55(11): 1866-1875.
https://doi.org/10.1038/s41588-023-01529-1
Parrish R.L., Gibson G.C., Epstein M.P., and Yang J., 2022, TIGAR-V2: efficient TWAS tool with nonparametric Bayesian eQTL weights of 49 tissue types from GTEx V8, Human Genetics and Genomics Advances, 3(1): 100078.
https://doi.org/10.1016/j.xhgg.2021.100068
Porcu E., Rüeger S., Lepik K., Santoni F.A., Reymond A., and Kutalik Z., 2019, Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits, Nature Communications, 10(1): 3300.
https://doi.org/10.1101/377267
Rasooly D., Peloso G.M., and Giambartolomei C., 2022, Bayesian genetic colocalization test of two traits using coloc, Current Protocols, 2(12): e627.
https://doi.org/10.1002/cpz1.627
Shikov A.E., Skitchenko R.K., Predeus A.V., and Barbitoff Y.A., 2020, Phenome-wide functional dissection of pleiotropic effects highlights key molecular pathways for human complex traits, Scientific Reports, 10(1): 1037.
https://doi.org/10.1038/s41598-020-58040-4
Tambets R., Kolde A., Kolberg P., Love M.I., and Alasoo K., 2024, Extensive co-regulation of neighboring genes complicates the use of eQTLs in target gene prioritization, Human Genetics and Genomics Advances, 5(4): 100187.
https://doi.org/10.1016/j.xhgg.2024.100348
Votava J.A., and Parks B.W., 2021, Cross-species data integration to prioritize causal genes in lipid metabolism, Current Opinion in Lipidology, 32(2): 141-146.
https://doi.org/10.1097/MOL.0000000000000742
Wainberg M., Sinnott-Armstrong N., Mancuso N., Barbeira A.N., Knowles D.A., Golan D., Ermel R., Ruusalepp A., Quertermous T., Hao K., Björkegren J.L.M., Im H.K., Pasaniuc B., Rivas M.A., and Kundaje A., 2019, Opportunities and challenges for transcriptome-wide association studies, Nature Genetics, 51(4): 592-599.
https://doi.org/10.1038/s41588-019-0385-z
Wallace C., 2021, A more accurate method for colocalisation analysis allowing for multiple causal variants, PLoS Genetics, 17(9): e1009440.
https://doi.org/10.1371/journal.pgen.1009440
Xie Y., Shan N., Zhao H., and Hou L., 2021, Transcriptome-wide association studies: general framework and methods, Quantitative Biology, 9(2): 141-150.
https://doi.org/10.15302/J-QB-020-0228
Zhang J., and Zhao H., 2023, eQTL studies: from bulk tissues to single cells, Journal of Genetics and Genomics, 50(12): 925-933.
https://doi.org/10.1016/j.jgg.2023.05.003
Zhang Y., Wang M., Li Z., Yang X., Li K., Xie A., Dong F., Wang S., Yan J., and Liu J., 2024, An overview of detecting gene-trait associations by integrating GWAS summary statistics and eQTLs, Science China Life Sciences, 67(6): 1133-1154.
https://doi.org/10.1007/s11427-023-2522-8
Zhao S., Crouse W., Qian S., Luo K., Stephens M., and He X., 2022, Adjusting for genetic confounders in transcriptome-wide association studies leads to reliable detection of causal genes, bioRxiv, 2022(9): 1-46.
https://doi.org/10.1101/2022.09.27.509700
Zheng Z., Huang D., Wang J., Zhao K., Zhou Y., Guo Z., Zhai S., Xu H., Cui H., Yao H., Wang Z., Yi X., Zhang S., Sham P.C., and Li M.J., 2020, QTLbase: an integrative resource for quantitative trait loci across multiple human molecular phenotypes, Nucleic Acids Research, 48(D1): D983-D991.
https://doi.org/10.1093/nar/gkz888
Zhu H., and Zhou X., 2020, Transcriptome-wide association studies: a view from Mendelian randomization, Quantitative Biology, 2020: 1-15.
Zuber V., Grinberg N.F., Gill D., Manipur I., Slob E.A., Patel A., Wallace C., and Burgess S., 2022, Combining evidence from Mendelian randomization and colocalization: review and comparison of approaches, The American Journal of Human Genetics, 109(5): 767-782.
https://doi.org/10.1016/j.ajhg.2022.04.001




. PDF(1564KB)
. FPDF(win)
. FPDF(mac)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Xuanjun Fang
Related articles
. Statistical genetics
. Causal inference
. Genome-wide association study (GWAS)
. Molecular QTL (eQTL, sQTL, pQTL)
. Transcriptome-wide association study (TWAS)
. Colocalization
. Mendelian randomization
. Multi-omics integration
. Pleiotropy
. Complex traits
Tools
. Email to a friend
. Post a comment